Inert Dusts/Hard metal pneumoconiosis:
Clinical:
Accumulation of non-fibrogenic dusts may also produce pulmonary parenchymal abnormalities. Particles are deposited at the end of conducting airways at their junction with the respiratory bronchioles. Conglomerations of these particles are radio-opaque, but there is no associated inflammatory response or fibrosis. Generally, these patients feel healthy and have no functional impairment, despite horrible looking chest radiographs. Some offending agents are iron (siderosis- electric arc and oxyacetylene welders may inhale finely divided particles of iron oxide), tin (stannosis), barium (baritosis), and antimony (antimony pneumoconiosis).
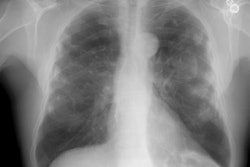
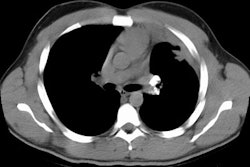
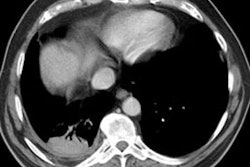
Siderosis occurs secondary to accumulation of iron oxide within pulmonary macrophages [2]. Due to admixture of the inhaled iron with silica, patients can develop silicosiderosis which may be associated with pulmonary fibrosis [2]. Typical radiographic findings include small nodules which are most prominent in the perihilar, middle third of the lungs [2]. HRCT shows small ill-defined centrilobular nodules (71% of patients) with or without fine branching structures (which indicate the deposition of minute iron oxide particles along perivascular and peribronchial lymphatic vessels [2]. Other findings include ground glass opacities, reticulation, and areas of high attenuation indicating organizing pneumonia with siderosis [2].
X-ray:
In general, CXR findings in patients with heavy metal pneumoconiosis typically demonstrate the presence of micronodules (small rounded opacities) which can mimic silicosis, but are most prominent in the perihilar regions. These changes are reversible and can resolve after exposure ceases. These nodules do not represent reactive fibrosis, but rather radiopaque accumulations of particles that lie within macrophages aggregated along the perivascular and peribronchial lymphatics.
HRCT will demonstrate the presence of ill-defined centrilobular micronodules [1]. Other findings include ground glass opacities
REFERENCES:
(1) Radiographics 2001; Kim KI, et al. Imaging of occupational lung disease. 21: 1371-1391
(2) Radiographics 2006; Chong S, et al. Pneumoconiosis: comparison of imaging and pathologic findings. 26: 59-77