MEN Syndromes:
MEN is a group of dominantly inherited disorders associated with
an increased susceptibility to endocrine and non-endocrine tumors
[5].
MEN I: (Wermer syndrome)
- Clinical:Autosomal dominant transmission (penetrance of at least 95%
secondary to a germline mutation involving the tumor suppressor
gene MEN1 which is located on chromosome 11q13 which codes
for the protein menin [1,4,5,7]. MEN1 has a prevalence of 2-20 per
100,000 patients [7].
The syndrome consists of:
1- Parathyroid lesions: Hyperplasia occurs more frequently than adenoma. Primary hyperparathyroidism occurs in 88-97% of cases and is usually the presenting feature [1,3]. Parathyroid adenomas occur in 65-90% of cases [5]. Treatment for hyperparathyroidism in these patients is typically surgery with removal of 3.5 of the 4 parathyroid glands [3]. Unfortunately, re-enlargement of the remaining parathyroid tissue can result in recurrent hyperparathyroidism in up to 67% of patients [3].
2- Pituitary tumors: Anterior pituitary adenomas occur in 30% (20-65% [4,5]) of MEN I patients [1]. Microadenomas are found in 50% of cases [5]. The tumors are usually functioning (60% secrete prolactin, less than 25% secrete growth hormone, and 5% secrete adrenocorticotropic hormone) [1].
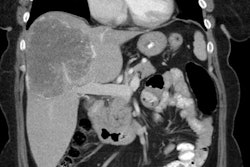
3- Pancreatic islet cell tumors: Endocrine tumors of the pancreas occur in 30-80% of patients [4,5]. Multiple islet cell tumors are common - these are generally small microadenomas [1]. Most pancreatic tumors in MEN I are functional with less malignant potential than sporadic pancreatic tumors [1].
Gastrinomas (producing Zollinger-Ellison syndrome) is the most common functioning pancreatic endocrine neoplasm in patients with MEN1 and account for 60% of cases [1,6,7]. In over half the cases these are multiple duodenal microgastrinomas (smaller than 5 mm) [1]. Up to 60% of patients with Zollinger-Ellison (ZE) syndrome are found to have MEN1 [7].
Insulinomas are the second most prevalent PNET in MEN1 and account for 10-30% of islet cell tumors in MEN I and coexist with gastrinomas in 10%, as well as with non-functioning tumors [1,4,6,7]. Patients with duodenal gastrinomas have a better prognosis than those that originate from the pancreas [4]. In addition to a functioning pancreatic endocrine tumor, patients with MEN1 often have several small coexisting mutlihormonal tumors that measure up to 5 mm- termed microadenomas [6].
Glucagonoma- Approximately 5-17% of glucagonomas occur in patients with MEN1 [7].
Cystic tumors constitute up to 15% of all PNETs in patients with MEN1 (typically large, likely benign, and mostly non-functioning [7].
Patients commonly present in the 3rd and 4th decade, but affects
a wide age range (5 to 81 years) and involves male and female
patients equally [2,7]. MEN I is also associated with carcinoid,
Cushing's disease, adrenal cortical lesions (between 16-40% of
patients with MEN I have adrenal cortical adenomas, which may
rarely be functional), lipomatous tumors, multiple facial
angiofibromas (seen in up to 85-90% of patients with MEN1) and
collagenomas [1,6,7].
Carcinoids develop in 2-5% of MEN I patients and these originate
almost exclusively in the foregut- thymus, bronchus, stomach, and
duodenum [1]. Thymic carcinoids in MEN I patients occur
predominantly in middle aged men [1]. The tumors usually behave
more aggressively than sporadic carcinoids [1]. Bronchial
carcinoids in MEN I patients are found more commonly in women [1].
There is a 30 fold increase in the prevalence of gastric
carcinoid in MEN I patients [1]. Of the three subtypes of gastric
carcinoids, type II is commonly identified in patients with MEN 1
[7]. They are commonly multicentric and metastasize to local nodes
and the liver [1]. Gastric carcinoids occur almost exclusively in
MEN patients with gastrinomas and are associated with
Zollinger-Ellison syndrome (affecting up to 40% of patients in
this group) [1]. Elevated gastrin levels in patients with ZE
syndrome produces hyperplasia of gastric enterochromaffin cells
that have a propensity to evolve into gastric carcinoids [7]. On
CT, when the stomach is distended with water, multiple small
gastric wall tumors can be identified during arterial phase
imaging [7].
MEN II:
The MEN 2 syndromes are due to a mutation of the RET proto-oncogene (locus on chromosome 10q11) which codes for the tyrosine kinase receptor [5,7].
MEN IIA: (Sipple syndrome)
- Clinical:Autosomal dominant disorder [1]. The syndrome consists of:
1- Parathyroid lesions: Hyperplasia or adenomas in 25% of patients [5].
2- Adrenal pheochromocytoma (often bilateral): Pheochromocytomas occur in 50% of MEN II patients [1]. The tumors are bilateral in up to 50% of cases (compared to 10% of sporadic tumors) [1].
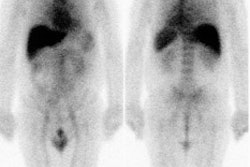
3- Medullary thyroid carcinoma: MTC occurs in nearly all patients with MEN II [1,5]. Patients are usually younger than those who develop sporadic MTC [1]. Local invasion is common and nodal spread to the neck and mediastinum occurs in up to 50% of cases [1]. Distant metastatic disease to the liver, lung, and bone occur in 15-25% of cases [1].
MEN IIA can also be associated with cutaneous lichen amyloidosis amd Hirschsprung disease (aganglianosis) [7].
MEN IIB (previously MEN III): (Mucosal Neuroma Syndrome)
- Clinical:A rare disorder, generally due to a new mutation with no family
history [1]. It is distinguished from MEN IIA by the lack of
parathyroid hyperplasia and the presence of non-endocrine features
(such as a marfanoid appearance and mucosal neuromas) [7].
It consists of:
1- Adrenal: Pheochromocytoma
2- Thyroid medullary carcinoma
3- Ganglioneuromatosis: Mucosal, oral and intestinal. Diffuse ganglioneuromatosis commonly affects the colon, terminal ileum, appendix, and stomach [7]. Patients most commonly present with constipation, but other symptoms include diarrhea and abdominal distention [7]. On contrast examination there is a prominent mucosal pattern with thickening of the bowel wall (thickening of the myenteric plexus and ganglia [7]) and colonic dilatation (megacolon [7]). The mucosa may resemble valvulae conniventes- "jejunalization of the colon," which is also seen in amyloidosis, jejuno-ileal bypass, and transplanted colon for colonic interposition.
4- Marfanoid appearance: Seen in all patients [1], but no ocular or cardiac findings
5- Prognathism, Melanosis, Myopathy, Thickened lips.
REFERENCES:
(1) Radiographics 2006; Scarsbrook AF, et al. Multiple endocrine neoplasia: spectrum of radiologic appearances and discussion of a multitechnique imaging approach. 26: 433-451
(2) Radiographics 2006; Chung EM, et al. Pancreatic tumors in chidlren: radiologic-pathologic correlation. 26: 1211-1238
(3) AJR 2008; Veldman MW, et al. Percutaneous parathyroid ethanol ablation in patients with multiple endocrine neoplasia type I. 191: 1740-1744
(4) Radiographics 2010; Lewis RB, et al. Pancreatic endocrine tumors: radiologic-clinicopathologic correlation. 30: 1445-1464
(5) Radiographics 2011; Monsalve J, et al. Imaging of cancer
predisposition syndromes in children. 31: 263-280
(6) AJR 2020; Davila A, et al. Multiple endocrine neoplasia:
spectrum of abdominal manifestations. 215: 885-895
(7) Radiographics 2020; Khanna L, et al. Pancreatic neuroendocrine neoplasms: 2020 update on pathologic and imaging findings. 40: 1240-1262