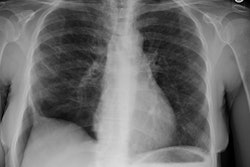
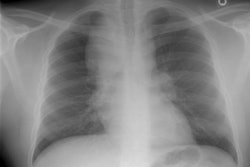
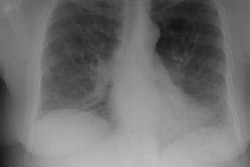
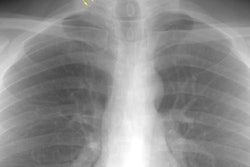
Drug Induced Lung Disease:
View cases of drug induced lung disease
Clinical:
Drugs, particularly cytotoxic agents, can result in a variety of lung injury including diffuse alveolar damage, interstitial pneumonitis, pulmonary edema, pulmonary hemorrhage, eosinophilic lung disease, BOOP, bronchiolitis obliterans, and pulmonary veno-occlusive disease. Four common drug-related processes in the lung are interstitial pneumonitis and fibrosis, hypersensitivity reaction, non-cardiogenic pulmonary edema, and bronchiolitis obliterans.
1. Interstitial pneumonitis and fibrosis:
Interstitial pneumonitis and fibrosis are typically associated with cytotoxic drugs such as bleomycin, methotrexate, and busulfan. Other agents, however, can also produce this type of injury including amiodarone, penicillamine, and nitrofurantoin. The radiologic findings are similar to UIP/NSIP/DIP, however, the abnormalities are typically bilateral and symmetric (as opposed to the patchy distribution of abnormalities associated with UIP/NSIP/DIP).
2. Hypersensitivity reaction:
Hypersensitivity reactions occur most commonly to methotrexate, nitrofurantoin, bleomycin, cyclophosphamide, and sulfonamides. The reaction is unrelated to the cumulative dose of drug received. On laboratory analysis, up to 40% of patients will demonstrate a peripheral eosinophilia. HRCT findings include patchy areas of ground glass attenuation or even consolidation. CT has been shown to be more sensitive in the detection of drug induced pulmonary toxicity (38% vs. 15% by CXR). Early there are areas of ground-glass attenuation, but with progressive damage, coarse reticular opacities and areas of consolidation develop [2].
3. Non-cardiogenic pulmonary edema/capillary leak syndrome: Non-cardiogenic pulmonary edema is associated with cytotoxic agents (mitomycin-C, cyclophosphamide, cytosine arabinoside [Ara-C], bleomycin, and methotrexate [5,11]), interleukin 2 [12], gemcitabine [12], aspirin, tricyclic antidepressants, and narcotics [11]. The onset is sudden, and usually within a few days of the onset of treatment. Transfusion-related lung injury also produces pulmonary edema and it is believed to be an immune-mediated event [11]. It generally occurs within 2 hours of a transfusion, but the reaction can be delayed out to 6 hours [11]. In the early stages, the CXR will show signs of pulmonary edema [11]. Over time the interstitial and alveolar infiltrates progress and a "white-out" appearance develops [11].
4. Bronchiolitis obliterans: Bronchiolitis obliterans is primarily associated with penicillamine and sulfasalazine agents [11].
5. Bronchiolitis oblietrans with organizing pneumonia (Cryptogenic organizing pneumonia): COP can occur secondary to bleomycin, nitrofurantoin, cyclophosphamide, methotrexate, amiodarone, and gold salts [11].
6. Pulmonary hemorrhage: Pulmonary hemorrhage is an unusual drug induced complication that can be seen with anticoagulant therapy, amphtericin B, Ara-C, mitomycin, penecillamine, and high dose cyclophosphamide [11]. Major and fatal hemoptysis can also be seen in up to 5% non-small cell lung cancer patients treated with bevacizumab [12].
7. Diffuse alveolar damage: DAD is a common manifestation of pulmonary drug toxicity. It is frequently caused by cytotoxic agents such as cyclophosphamide, bleomycin, and carmustine.
Specific Drug Induced Hypersensitivity Reactions:
Clinical:
Mechanisms of pulmonary injury associated with certain drugs include direct toxicity or a hypersensitivity reaction. Symptoms include the insidious onset of dyspnea, cough, and decreased lung volumes and diffusing capacity on pulmonary function testing. Some drugs associated with hypersensitivity reactions include:
1- Chemotherapeutic agents:
a) Bleomycin:
Bleomycin is the chemotherapeutic agent most commonly associated with pulmonary toxicity. Bleomycin is concentrated in the lung- as a result, some degree of pulmonary toxicity develops in about 4-20% of treated patients [3,4]. Diffuse alveolar damage is the most common manifestation of lung injury for this agent [7]. Doses in excess of 400 mg are associated with an increased risk for pulmonary toxicity [1,3]. The frequency and severity of toxicity increase with age, cumulative dose, and reinstitution of therapy within 6 months of discontinuation [3,4,7]. Lung damage is potentiated by concurrent radiation therapy or oxygen administration. The pulmonary abnormalities are also not confined to the radiation port, but may involve the entire lung [3]. There also appears to be a synergistic effect between bleomycin and cyclophosphamide [4]. The onset of clinical symptoms usually occurs 4 to 10 weeks after treatment. Patients complain of dyspnea, non-productive cough, and fever. Diffusing capacity for cardon monoxide (DLCO) is decreased and lung volumes are usually decreased as well. The course of bleomycin toxicity is variable. With mild toxicity discontinuation of the agent may lead to reversal of the abnormalities. In patients with pulmonary fibrosis the prognosis is poor, but a trial of steroids is warranted.
On chest radiographs, bibasilar reticular or fine nodular opacities are the earliest findings in bleomycin toxicity. With more severe involvement, these opacities progress to involve the middle and upper lung zones. The presence of multiple pulmonary nodules has also been reported with bleomycin toxicity (especially in association with a BOOP-type reaction [6,7]) and can mimic metastatic disease [2]. Rarely, patients may experience an anaphylactic response to the agent which can lead to pulmonary edema- treatment with steroids is more effective in these patients [3]. CT can detect parenchymal findings indicative of pulmonary toxicity prior to plain film findings [3].
b) Busulfan:
A dose greater than 500 mg is associated with a 4-10% incidence of pulmonary toxicity and concurrent radiation therapy increases the risk. Toxicity occurs from one month to 12 years after administration of the agent. Diffuse alveolar damage is the most common manifestation of busulfan-induced lung injury. Prognosis is poor. [6]
c) Carmustine (BCNU):
Carmustine is a nitrosurea compound used primarily to treat CNS malignancies with an incidence of pulmonary toxicity between 20-30% of treated patients. Pulmonary toxicity is dose related (cummulative dose greater than 1500 mg/m2 is associated with a 50% incidence of pulmonary toxicity [7]). Increased risk for toxicity is associated with underlying lung disease, prior thoracic radiation, and combination chemotherapy with cyclophosphamide. Patients present with progressive dyspnea on exertion and dry cough. The overall mortality from pulmonary toxicity is greater than 90% [3]. Radiographic abnormalities are usually not detected until patients have been symptomatic for some time. Typically there is bibasilar interstitial fibrosis [3], although other authors claim that diffuse alveolar damage is the most common lung injury associated with this agent [6,7].
d) Cyclophosphamide:
The incidence of cyclophosphamide pulmonary toxicity is rare. Diffuse alveolar damage is the most common manifestation of cyclophosphamide induced lung disease [7]. Symptoms may begin 2 weeks to 13 years following treatment. There is no relationship between development of lung injury and dose or duration of administration [6,7]. Patients present with dyspnea on exertion, cough, and fever. Treatment is with discontinuation of the agent and steroids with good to variable response. Overall mortality is about 50%. Radiographic findings are of basilar interstitial disease [3]. Diffuse alveolar damage can also occur with the use of this agent [6].
e) Cytosine arabinoside:
Also known as Ara-C, it is an antineoplastic agent. Acute pulmonary toxicity has been reported after treatment with intermediate or high dose levels. Patients complain of fever, dry cough, dyspnea, and hypoxemia. Symptoms usually develop during the course of administration of the agent, but may be delayed for close to one month. The chest radiograph may initially show apredominantly basilar interstitial or mixed interstitial-alveolar pattern. This progresses to a diffuse alveolar pattern in most cases which improved rapidly over 3 to 7 days. Pathologically, there is pulmonary edema with a highly proteinaceous interstitial and intra-alveolar infiltrate. [3]
f) Interleukin-2:
IL-2 stimulates production of circulating lymphokine-activated killer cells. IL-2 is associated with a high incidence of toxic reactions including non-cardiogenic pulmonary edema (likely the result of a leaky capillary syndrome). Radiographic findings can range from a mild interstitial edema to frank alveolar edema. The findings generally occur within 2 to 8 days of initiation of therapy and resolve within 4 days of termination of treatment. [3]
g) Methotrexate:
The incidence of pulmonary toxicity in patients receiving methotrexate between 5-10% [7]. The toxicity is NOT dose related, although patients that receive treatment more frequently (ie: daily or weekly) may be more susceptible to lung injury [3]. Patients generally present with fever, chills, malaise, headache, cough, and dyspnea within weeks of initiation of therapy. Peripheral eosinophilia is found in about 50% of cases and a skin eruption is seen in up to 17% of patients. The prognosis is good and the toxicity usually regresses after cessation of the agent. Radiographic findings usually clear over time (although pulmonary fibrosis may develop) [3]. Overall mortality from pulmonary toxicity is 10%.
Methotrexate toxicity begins as an interstitial pattern (reticular or reticulo-nodular), but can present with air space consolidation. The pattern can be primarily nodular. Associated adenopathy (10%) or pleural effusion can be seen. Rarely patients may present with acute pleuritis (associated with the presence of a pleural effusion) or non-cardiogenic pulmonary edema (following intrathecal administration of the agent).
h) Thalidomide:
Thalidomide is an immunomodulator used in the treatment of multiple myeloma that is refractory to other chemotherapy [16]. The agent is associated with an increased incidence of thromboembolic disease especially when combined with dexamethasone [16]. The median time of onset of a thrombotic event is approximately 3 months following intiation of therapy [16].
i) Targeted Agents:
Click here for a list of commonly used targeted therapy agents and their toxicities
Targeted agents for malignancies exploit a key signal pathway for their therpaeutic effect and include both monoclonal antibodies and small molecules [13]. Monoclonal antibodies (typically ending in "-mab") target receptor molecules on the surface of cells, whereas small molecules (typically ending in "-ib") can penetrate the cell emembrane and interact either with the intracellular domain of a transmembrane receptor molecule or with deep targets inside a cell [13]. Monoclonal antibodies tend to be failrly specific- targeting only one receptor or a narrow family or receptors, whereas small molecules can interact with multiple intraceullar targets [13]. Agents with primary molecular targets on the cell surface include epidermal growth factor receptor (EGFR) inhibitors (monoclonal antibody agents cetuximab and panitumumab; and the small molecule recptor tyrosine kinase inhibitors erlotinib [Tarceva] and gefitinib); human ErbB2 (formally HER2) receptor inhibitors (trastuzumab, pertuzumab, lapatinib); vascular endothelial growth factor (VEGF) and VEGF receptor inhibitors (bevacizumab, aflibercept, axitinib, pazopanib, sorafenib, sunitinib); KIT/tyrosine kinase inhibitors (or stem cell factor receptor) inhibitors (imatinib [Gleevec], sunitinib); and the anaplastic lyphoma kinase (ALK) inhibitor crizotinib [13]. Agents with molecular targets downstream of cell surface receptors include RAF inhibitors (sorafenib, vemurafenib) and the mammalian target of rapamycin inhibitors (temsirolimus, everolimus) [13].
Antiangiogenic molecular targeted therapies produce morphologic changes in tumors such as decreased enhancement and attenuation with or without concurrent changes in size [17]. As such, RECIST criteria can underestimate treatment response [17].
Epidermal growth factor (EGFR) inhibitors:
Cetuximab and panitumumab are indicated for the treatment of colorectal cancer with absence of a downstream KRAS mutation (which if present renders EGFR inhibition therapy ineffective) [13]. Cetuximab is also indicated for treatment of squamous cell carcinoma of the head and neck [13]. Erlotinib is indicated for treatment of non-small cell lung cancer and metastatic pancreatic cancer [13]. Gefitinib has limited approval for use in NSCLC [13].
Osimertinib is a third generation EGFR tyrosine kinase inhibitor that targets T790M second-site EGFR that is responsible for acquired resistance to conventional EGFR inhibitors [19]. A novel type of drug-related pulmonary phenomenon called transient asymptomatic pulmonary opacities has been described in up to 20% of patients with NSCLC treated with osimertinib [35]. Patients develop localized pulmonary opacities that resolve without any treatment during continued osimertinib therapy with a median 6 week duration [35]. Interestingly, patients who develop this apparent grade I pneumonitis had longer progression free and overall survival compared to patients without transient asymptomatic pulmonary opacities [35].
Pulmonary infiltrates and ILD are among the most common adverse reactions to EGFR inhibitors such as gefitinib and erlotinib [12,13]. The risk is highest in smokers and in patients with pre-existing lung disease [13]. The overall prevalence of pulmonary toxicity with gefitinib is 1% and is higher in Asia (4-5%) than in the US [12,19]. Clinically patients present with acute onset dyspnea and symptoms can appear as early as 1 week to 1 month following initiation of therapy [12]. CT findings include airspace consolidation or exensive bilateral ground glass infiltrates [12].
HER2 inhibitors
HER2 is a transmembrane tyrosine kinase receptor belonging to the EGFR family that regulates cell growth, survival, adhesion, migration, and differentiation [19]. Approximately 20% of patients with breast cancer have HER2 overexpression [19]. HER2 inhibitors include trastuzumab, lapatinib, pertuzumab, and trastuzumab emtansine (an immunoconjugate that combines trastuzumab with the microtubule targeting agent emtansine) [19].
Agents targeting ErbB2
These include the monoclonal antibody agents trastuzumab (Herceptin) and pertuzumab, as well as the tyrosine kinase inhibitor lapatinib [13]. All three agents are indicated for the treatment of breast cancers that overexpress ErbB2 [13]. Cardiotoxicity is the most relavant toxic effect of these agents and is most common with trastuzumab [13].
Agents targeting VEGF/VEGFR (Antiangiogenic therapies)
Vascular endothelial growth factor (VEGF) is an appealing antiangiogenic target because drugs unlike cancer cells ,endothelial cells are genetically stable and less likely to become treatment-resistant [17]. Anti-VEGF molecular targeted therapies are associated with striking intra-tumoral density changes [24].
Bevacizumab is a monoclonal antibody antiangiogenic agent that binds to free VEGF in the circulation and reduces its available concentration for binding to VEGF receptors [16,17]. This reduction leads to a diminished blood supply to the tumor and tumor shrinkage through downregulation of the VEGF axis [16]. Bevacizumab is indicated for the treatment of colorectal cancer, NSCLC of non-squamous histologic type (because of a risk of life-threatening pulmonary hemorrhage in squamous cell lung cancer, especially with central or cavitating tumors), and renal cell carcinoma [13,17].
Aflibercept is approved for colorectal cancer [13]. Axitinib is used for renal cell carcinoma [13]. Pazopanib is used for renal cell carcinoma and advanced soft tissue sarcomas [13]. Sorafenib and Sunitinib are tyrosine kinase inhibitors [14]. Sorafenib is used for renal cell carcinoma and hepatocellular carcinoma [13]. Sunitinib is used for renal cell carcinoma, GIST, and pancreatic neuroendocrine tumors [13]. Complications of these agents are related to vascular endothelial cell disruption [13].
Pneumatosis and bowel perforation are associated with use of these agents due to interference with bowel microvasculature leading to ischemia and thrombosis of vessels [13,14,24]. Pneumatosis can occur early after initiation of treatment and up to 71% of patients can be asymptomatic [17].
Bevacizumab is associated with an approximately 0.9-4% risk for bowel perforation (however, it has been reported to be as high as 10% in patients with tumor seeding serosal surfaces) [13,14]. The risk for perforation is higher in patients receiving higher doses [14] and in patients with colorectal and renal cancers [20]. Perforation usually occurs within the first 6 months of treatment [14]. These agents should be used with caution in patients with serosal implants, implants adjacent to bowel, in patients with pre-existing ulcerations, and in patients with weak bowel walls from an underlying condition such as Crohn disease, UC, or tumor necrosis [14]. Most patients can be managed conservatively with prompt discontinuation of the agent [17]. It is usually recommended to discontinue bevacizumab at least 6 weeks before elective surgery as it interfers with healing and increases the risk for anastomotic dehiscence [17].
Gastrointestinal fistulas and nongastrointestinal fistulas have been reported including tracheoesophageal, bronchopleural, biliary, vaginal, renal, and vesical [13].
Hemorrhage can occur with these agents and can be minor or serious in nature [13]. VEGF plays an important role in maintaining the integrity of endothelial cells [16]. It is postulated that inhibition of VEGF leads to a decrease capacity to renew endothelial cells, which makes vessels weak and prone to hemorrhage [16]. Mild bleeding episodes can be seen in up to 40% of patients receiving Bevacizumab [16], but potential life-threatening hemorrhage may occur in patients with squamous cell carcinoma who receive bevacizumab. In NSCLC, pulmonary hemorrhage has been associated with squamous cell histology, disease location close to major blood vessels, and with baseline tumor cavitation or necrosis [13,19].
Acute pancreatitis is a rare complication reported with pazopanib, sorafenib and sunitinib, although asymptomatic elevations of serum amylase and lipase may occur in up to 50% of patients treated with VEGF/VEGFR pathway tyrosine kinase inhibitors [13,17]. Gallbladder complications range from asymptomatic gallbladder distention and edema to acute acalculous cholecystitis [17].
Arterial thrombotic events (not venous) can occur including CVA and MI [13]. In several studies, Bevacizumab has been associated with an increased risk for venous thromboembolic disease [16,49]. In colon cancer patients receiving bevacizumab, PE can occur in up to 19% of patients [19].
Pneumonitis can be seen in 1.1% of patients treated with EGFR inhibitors [49]. Transient asymptomatic opacities (TAO) is a milder form of pneumonitis in patients treated with EGFR-TKIs (in particular osimertinib) [49]. TAO demonstrates new focal opacities on chest CT in asymptomatic patients, often with a simple pulmonary eosinophilia pattern, and the opacities typically resolve in 6-10 weeks without treatment [49].
A progessive reversible encephalopathy syndrome (PRES) is an uncommon (<0.1%) complications of VEGF/VEGFR therapy that has been reported with bevacizumab sorafenib and sunitinib [13]. The syndrome manifests as HA, nausea, seizures, and visual loss [17]. Imaging findings in PRES include cortical and subcortical white matter abnormalities/edema in the occipital, posterior temporal, and parietal lobes [17].On MRI, the condition manifests as T2-hyperintense lesions in the posterior circulation, mostly involving subcortical white matter, but with occasional invovlement of the overlying cortex [13].
Osteonecrosis of the jaw - these agents (including bevacizumab and sunitinib) may increase the risk for osteonecrosis of the jaw when administered in combination with bone modifying agents [13].
Thyroid dysfunction, especially hypothyroidism, has been reported to occur with sunitinib, sorafenib, pazopanib, and axitinib with an incidence as high as 90% in some studies [17].
Agents targeting KIT/tyrosine kinase inhibitors
Imatinib (Gleevec) and sunitinib are indicated for treatment of GIST [13]. Fluid retention (ascites or pleural effusion) and systemic body edema can occur as a result of the treatment [13]. Fluid retention with imatinib ranges from mild to severe and occurs in up to 74% of patients [20]. Hemorrhage with imatinib is especially prevalent with bulky GISTs, occuring in up to 5% of patients [13]. The hemorrhage is predominantly intra- or peritumoral [13]. Imatinib also causes elevation in serum liver enzymes in 15-20% of patients [13]. Imatinib is also associated with interstitial pneumonia and ILD [15]. Dasatinib is used for treatment of chronic myelogenous leukemia and pleural effusions can occur in 10-35% of patients treated with the agent and a concomitant pericardial effusion is often seen [15]. Dasatinib can also induce severe precapillary pulmonary hypertension [15].
Agents targeting anaplastic lymphoma kinase (ALK inhibitors)
The ALK-rearranged oncogene is present in 2-7% of patients with NSCLC [19]. Crizotinib is used for treatment of a subset of NSCLC containing a fusion oncogene rearrangement [13]. Severe pneumonitis can occur in 1-2% of patients usually wihtin 2 months of treatment initiation [13]. Patients present with dyspnea, with or without cough [13]. Other agents include alectinib and ceritinib [19].
Agents targeting RAF
These include sorafenib and vemurafenib (which is used in the treatment of melanoma with a BRAF mutation) [13]. Vemurafenib may induce a nodular subcutaneous panniculitis- these nodules can demonstrate activity on FDG PET imaging [13]. Vemurafenib is also associated with development of cutaneous squamous cell carcinomas in up to 24% of patients which can also be FDG avid [13].
Agents targeting mammalian target of rapamycin (mTOR)
mTOR is a serine/threonine protein kinase and is a critical component of the phosphatidylinositol 3-kinase pathway (PI3K)/Akt/mTOR pathway [19]. The agents include the serine-threonine kinases temsirolimus and everolimus which are both indicated for treatment of renal cell carcinoma [19]. Everolimus is also indicated for treatment of hormone-receptor positive HER-2 negative breast cancer, pancreatic neuroendocrine tumors, angiomyolipoma, advanced pancreatic neuroendocrine tumors, and subependymal giant cell astrocytoma associated with tuberous sclerosis [13,19].
Both agents are associated with pneumonitis with an incidence of about 30% among patients with RCC, 25% in NSCLC, and 21% in patients with advanced neuroendocrine tumors [19,35]. In one series, 36% of patients taking temsirolimus developed pulmonary abnormalities- either GGO with or without interstitial disease and parenchymal consolidation [13]. Asymptomatic cases can be monitored while continuing therapy [19]. In symptomatic cases, therapy should be withheld and corticosteroids may be required [19].
Targeted immune modulators/ Immune checkpoint inhibitors
Depending on the number of mutations in cancer cells, immune cells are able to recognize and attack them [31]. However, tumors employ several mechanisms to evade attack by the immune system with coinhibitory signaling leading to downregulation or "switching off" cytotoxic effector T-cell function [21,31,42]. One way tumor cells accomplish this is through up-regulation of inhibitory immune checkpoint pathways, such as the cytotoxic T-lymphocyte antigen-4 (CTLA-4), the programmed cell death protein (PD-1), or the programmed cell death 1 ligand (PD-L1) which are expressed by T-cells [21,31]. These pathways function to down-regulate T-cell effector function [31]. Tumors can exploit these checkpoint pathways by expressing co-inhibitory proteins to block anti-tumor immune responses, leading to an anergic T-cell phenotype, evasion of immune surveillance, and subsequent avoidance of immune attack [22,23,31].
Specifically, CTLA-4 is a protein receptor located on T-cell membranes which limits T-cell activation by raising the activation threshold and attenuating clonal expansion of anti-tumor specific T-cells [21,34]. An increase in CTLA-4 activity reduces immune response to tumor antigens [41]. PD ligands act as a brake on effector T-cell function [21,34]. PD-1 is a cell surface protein similar to CD28, with inhibitory effects on immune response [41]. Tumor-specific T-cells and tumor infiltrating lymphocytes have high PD-1 expression, resulting in decreased T-cell function with impaired cytokine production and upregulation in the tumor microenvironment [41].
Immune checkpoint inhibitors are monoclonal antibodies directed against one of these immune checkpoint pathways to inhibit the immune escape mechanism- thereby reactivating the immune system to recognize and attack tumor cells [18,31,39]. By releasing the immune system from inhibitory signals released by the tumor, these agents can promote tumor elimination [18]. Anti-CTLA-4 agents primarily invoke T-cell priming and expansion in lymph nodes [30].
Anti-PD1 and PD-L1 agents act predominantly within the tumor micro-environment by blocking immunosuppressive interaction between PD-1 on T cells and PD-L1 on cancer cells [30]. PD1 is expression in malignancies can be associated with a poor prognosis [22]. On immunohistologic analysis of tumors, PD-L1 expression is associated with clinical response to immune-checkpoint inhibitors (making it a biomarker for patient selection for treatment) [34]. In NSCLC, tumors with less than 1% PD-L1 expression are unlikely to respond to anti-PD-1 of anti-PD-L1 treatment [42]. Tumors with 1%-49% expression are more likely to respond when anti-PD therapy is combined with cytotoxic chemotherapy [42]. Tumors with more than 50% expression are likely to respond to anti-PD therapy alone [42]. However, PD-L1 immunohistochemistry is imperfect and up to 10% of "non-expressers" can respond to treatment [42]. This is because PD-L1 expression can be heterogeneous both in tumors and between different tumors in the same patient over time (therefore, biopsy specimens provide only limited information about the tumor phenotype) [34]. Tumors with a high tumor mutational burden, which refers to the number of somatic mutations found by DNA sequencing of a tumor specimen, may be more likely to respond to ICI therapy [44].
PD-1 inhibitors include nivolumab, pembrolizumab, cemiplimab, and dostarlimab [44]. PD-L1 inhibitors include atezolizumab, avelumab, and durvalumab [44].
Anti-PD-L1 atezolizumab has been labeled with 89Zr or 64Cu to produce PET tracers that can be used to document PD expression in tumors [42].
Tumor response:
Atypical response patterns can occur and are more common in immunogenic malignancies such as malignant melanoma and less frequent in other solid cancers [42].
Pseudoprogression- A subset of patients treated with immune checkpoint inhibitors manifest an atypical response with an apparent initial increase in tumor burden (with new or enlarging lesions or increased tumor metabolism on PET imaging), eventually followed by a partial or complete response of stable disease [18,21,33,41]. This atypical pattern has been attributed to tumor infiltration with inflammatory immune cells and/or continued tumor growth while awaiting development of an adequate immune response [18]. According to iRECIST if a target lesion increases in size by more than 20% or if new lesions appear, the treatment response should be classified as "unconfirmed progression of disease" [49]. Therefore in order to accurately confirm lack of progressive disease, an increase in total tumor burden must be identified on two consecutive studies performed at least 4 weeks apart [18,21]. If the subsequent CT shows further growth of the target lesion, growth of the new lesion, an increase in size of any nontarget lesions,or if new lesions appear, then the response pattern should be classified as "confirmed progression" [49]. In two-thirds of cases, pseudoprogression is observed before week 12 (early), whereas in one-third of cases it occurred after week 12 (delayed) [30]. Continued use of immune checkpoint inhibitors is beneficial in patients with pseudoprogression, especially if their clinical course appears to be stable or improving [48].
Pseudoprogression is seen in 2-14% of patients- overall average incidence of 6% and the incidence varies according to the type of cancer [18,21,33,42] (hence, most cases with progression at followup scanning likely represent true progression [48]). It can occur in 5-18% of patients with melanoma, up to 7% of patients with non-small cell lung cancer, 5-7% of renal cell carcinomas, and 1-2% in head and neck cancers [41,42,47]. The type of immune checkpoint inhibitor can also impact the observed rate of pseudoprogression which has been reported to be more commonly observed with CTLA-4 therapy (10% incidence), than with anti-PD-1 monoclonal antibody therapy (6%) [21,30,42]. Pseudoprogression associated with the anti-PD-L1 agent atezolizumab is more common with melanoma patients (7.3%), compared to lung cancer patients (2.3-2.8%) [30].
In one study of the anti-PD-1 agent pembrolizumab in patients with advanced melanoma, pseudoprogression was reported in 7% of patients and was associated with an improved 2 year survival compared to non-responders [42].
Dissociated response: Dissociated response is defined as the coexistence of responding and non-responding lesions (a mixed metabolic response on PET imaging) and likely reflects the heterogeneity of the tissue-specific tumor microenvironment and the emergence of treatment-induced resistant tumor clones [41,42,48]. It can be seen in up to 10% of patients [41]. It is overall associated with a favorable outcome and patients with dissociative response and oligoprogession benefit from continuation of ICI therapy and local ablative treatments to growing lesions [41].
Hyperprogressive disease (HPD): In a subset of patients, there is growing evidence that treatment with immune checkpoint inhibitors can have a detrimental effect characterized by a rapid increase in tumor extent and a worse clinical outcome- this is referred to as hyperprogressive disease [29]. Common definitions include a two-fold or greater increase in tumor volume growth rate during immunotherapy, time to treatment failure of less than 2 months, and clinical deterioration according to the Eastern Cooperative Oncology Group [41]. So far- this appears to be associated with anti-PD-1 and anti-PD-L1 therapy [30]. The prevalence of HPD varies from 4% to 29% [30,47] and if present requires immediate cessation of checkpoint inhibitor treatment and a switch to an alternate therapy [48]. The etiology for hyperprogression is not well understood, but may be related to reprogramming of tumor-associated macrophages through interaction with anti-PD-1 [30]. In NSCLC, patients with high metabolic tumor burdens on PET imaging (expressed by MTV and TLG), may be at increased risk for HPD following initiation of immune checkpoint inhibitor therapy [29]. Other factors that may be associated with an increased risk for HPD include age over 66 years and female sex [29].
Unfortunately, immune checkpoint blockade can lead to induction of hyperactivated T-cell-mediated responses against normal tissues [18]. These agents can produce a variety of complications including dermatologic rash and erythema, hypophysitis, thyroiditis (up to 22% of patients), uveitis, pneumonitis, hepatitis, pancreatitis, and colitis [19,21,28,42]. It has been reported that CTLA-4 therapy has a higher incidence of immune adverse events (54-90%), compared to anti-PD-1 (26%) and anti-PD-L1 (13.7%) agents [21,28]. Adverse event risk has been shown to have a dose dependent relationship with CTLA-4 inhibitors, but this has not been consistently observed with PD-1 or PD-L1 inhibitors [28]. Combinations of PD-1 and CTLA-4 inhibitors have demonstrated higher rates of immune adverse events compared to monotherapies in patients with advanced melanoma [28]. Pneumonitis and thyroid disorders more commonly occur with PD-1/PD-L1 therapy, whereas hypophysitis and colitis more commonly occur with anti-CTLA-4 [41]. The majority of immune adverse events occur in the induction phase, usually within the first 12 weeks of initiation of therapy, although late events manifesting after one year have also been observed [28]. Click here to view a summary of thoracic complications associated with targeted cancer therapies [19].
Immune adverse events:
The combined use of ipilimumab and nivolumab often results in a higher incidence of adverse events than monotherapy [44]. However, some studies suggest that patients who develop immune adverse events have superior progression free and overall survival compared to patients that do not develop adverse events [44]. Treatment of immune adverse events usually consists of glucocorticoids and, if necessary, suspending or discontinuing immunotherapy [44].
Pneumonitis: Pneumonitis can occur in 3-6% of patients treated with immune checkpoint inhibitors [28]. Higher rates of pneumonitis have been observed in patients with non-small cell lung cancer and renal cell carcinoma, compared to those with melanoma [28]. In non-small cell lung cancer patients, the incidence and severity of pneumonitis has been shown to be higher in those receiving PD-1 inhibitors compared to PD-L1 inhibitors (3.6% vs 1.3%) [28,35]. The risk is also higher in patients receiving combination therapy compared with monotherapy (up to 10% in patients receiving combination PD-1/PD-L1 and CTLA-4 inhibitors [41]) [35]. Epidermal growth factor receptor inhibitors given concurrently with ICI therapy or after prior ICI therapy have been shown to lead to higher rates of pneumonitis [44]. A higher incidence of pneumonitis has also been noted in patients with a history of asthma or COPD (5.3%) and in patients with a history of thoracic radiation (6%) [35]. A significant correlation between anti-PD-1-related pneumonitis risk and preexisting pulmonary fibrosis has also been reported [43]. Other risk factors include Asian ethnicity, former or current smoking, and prior chest radiation therapy [44]. Pneumonitis is the leading cause of PD-1 inhibitor and PD-L1 inhibitor-related deaths [44].
The time to onset of pneumonitis is variable- ranging from 9 days to 19 months after treatment (median time of onset is 2.8 months- typically 10-12 weeks) [28,41]. The onset has been shown to occur earlier in lung cancer patients compared to those with melanoma (2.1 months versus 5.2 months, respectively) [28].
The most common presenting symptoms are dyspnea and cough, but up to one third of patients may be asymptomatic at onset [28].
Various patterns of pneumonitis can be observed- organizing pneumonia (most common), NSIP (next most common), hypersensitivity pneumonitis, AIP-ARDS, and bronchiolitis (tree-in0bud) [28]. A sarcoid-like reaction can also be seen, and has been associated with an eventual therapeutic response in melanoma patients [28].
Treatment for therapy-related pneumonitis is based on symptom severity consisting of withholding the agent and initiation of steroid or immunosuppressive therapy [28,41]. In general, about one-third of patients that develop pneumonitis are able to restart therapy after successful treatment of the pneumonitis [32]. Despite successful treatment, about one-fourth (25-28%) of patients will develop recurrence following retreatment and this usually has an earlier onset [28,32] and a similar, but more extensive imaging pattern than the initial pneumonitis [44]. It is often safe to restart immunotherapy in patients with grade 1 or 2 pneumonitis (recurrence rate is 17-29%) [28]. Patients initially diagnosed with grade 3 or 4 pneumonitis, generally discontinue therapy permanently [28].
Myocarditis: ICI induced myocarditis most likely occurs (80% of cases) within the first 3 months after treatment initiation (median time of onset is 30 days) [36,44]. It is uncommon (although it is the most common cardiovascular adverse event associated with ICI treatment [44]), but it is one of the most serious immunotherapy-related adverse events with a fatality rate of 30-50% [40]. Corticosteroid therapy is required to improve prognosis [40]. Sub-clinical ICI cardiotoxicity may be more common than previously assumed and can be detected by cardiac MR imaging [36]. Findings on MRI include myocardial edema and late gadolinium enhancement with a variable pattern (including subendocardial, subepicardial, mid-myocardial, and transmural) [44].
Enterocolitis: Colitis is the second most common adverse effect after cutaneous involvement and is the most common cause of death among patients with immune adverse events [41]. It generally occurs 5-10 weeks after initiation of therapy [41] (other authors indicate a median time to onset of 5 weeks with anti-CTLA-4 therapy and 2-4 months with anti-PD-1 therapy [44]). The incidence of severe colitis is higher with ipilimumab containing treatment regimens (up to 7%) and with combination therapy (ipilimumab and nivolumab- up to 9%) [44]. Severe colitis is seen in about 1% of patients treated with anti-PD-1 or anti-PD-L1 therapy [44]. Involvement can be diffuse (most common- 75% of cases), segmental, and isolated rectosigmoid [41].
Sarcoid-like reaction/lymphadenopathy: A rare sarcoid-like reaction can occur 3-36 weeks (approximately 3 months) after initiation of therapy [41,44]. Sarcoid-like reactions are most common with the anti-CTLA-4 agent ipilimumab (5-7% incidence) and less commonly with anti-PD-1 and anti-PD-L1 agents [44]. Sarcoid-like reaction tends to be less severe than pneumonitis and may not require discontinuation of ICI therapy [44]. Most commonly the reaction results in bilateral mediastinal and hilar adenopathy with increased tracer uptake on FDG PET imaging [42].
Thyroiditis: Thyroid dysfunction is mainly associated with anti-PD-1 therapy and combined anti-PD-1 and anti-CTLA-4 therapy [44]. The condition most commonly manifests as painless thyroiditis and the incidence is about 6.5% [44]. The median time of onset is 5.3 weeks from initiation of therapy [44]. Patients usually present with overt or subclinical thyrotoxicosis [44].
Hypophysitis: Hypophysitis occurs in up to 10% of patients on anti-CTLA-4 therapy- either with monotherapy or slightly more commonly with combine anti-PD-1 therapy [44]. The condition is much less common with anti-PD-1 or anti-PD-L1 monotherapy (0.5-1%) [44]. The median time of onset is 9-13 weeks after treatment initiation with ipilimumab (alone or in combination) and 26 weeks after initiation of anti-PD-1 therapy [44].
Impilimumab:
Impilimumab is an immune modulator used for the treatment of metastatic melanoma that functions by inhibiting the cytotoxic T-lymphocyte antigen-4 (CTLA-4 inhibitor) mediated negative regulatory signals, thus boosting the host anti-tumor response by promoting T-cell activation [13,18]. The agent interacts with the (CTLA-4) rceptor on the surface of T-cells which prevents ligand binding that would otherwise down-regulate T cell function [13]. The absence of the inhibitory signal is thought to promote T-cell mediated antitumor activity [13].
Impilimumab is associated with grade 3 or 4 enterocolitis in 10-20% of patients [13] and diarrhea and colitis typically occur 6-7 weeks after initiation of treatment [21]. The colitis can be diffuse (75% of cases) or segmental (and associated with underlying diverticulosis) [27].
Hypophysitis can occur in 4-13% of patients with a median time of 8.4 weeks from initiation of therapy [13,21,27]. Patients with hypophysitis present with HA, weakness, and fatigue and develop anterior hypopituitarism with multiple hormonal deficiencies [21,27]. MR will demonstrate mild-to-moderate diffuse pituitary enlargement and thickening of the pituitary stalk is common [21]. Treatment for hypophysitis is systemic high dose corticosteroids [27].
The agent can also induce benign lymph node enlargement (a sarcoid-like granulomatosis and lymphadenopathy involving the chest has been reported in 5-7% of patients with melanoma [19,20]) and inflammatory soft tissue changes including myositis, fasciitis, and retroperitoneal fat haziness [13]. Other adverse effects include dermatitis (18-68% of patients), hepatitis (3-9%), and pneumonitis (5%) [20,21].
PD-1 inhibitors:
PD-1 is a negative co-stimulatory receptor expressed on the surface of activated T and B cells [23]. Tumor cells can avoid destruction by the host immune system by producing the PD-L1 ligand [20]. PD-L1 present on the surface of tumor cells interacts with the PD-1 protein present on the surface of T-cells and B-cells [20]. Binding of the PD-L1 ligand to PD-1 causes downregulation of T-cell activity (lowering the immune response) and allows tumor cells to evade destruction [20]. Monoclonal antibodies targeting PD-1 receptors on T-cells have been developed to inhibit the downregulation of T-cell activity by tumors and to promote the body's innate antitumor response [20]. Current agents include nivolumab and pembrolizumab to treat melanoma [20]. Adverse effects include maculopapular skin rash, arthritis (which can persist for more than 3 months following cessation of therapy), colitis, pneumonitis, and endocrine and hepatic toxic effects [20,26].
Nivolumab is a programmed death-1 inhibitor (PD-1 inhibitor) that has been shown to result in improved survival in patients with NSCLC [18]. The agent has also been used to treat melanoma, renal cell carcinoma, urothelial carcinoma, and Hodgkin lymphoma [19]. Immune related adverse events can occur in 32-58% of patients [18]. The incidence of pneumonitis for nivolumab/pembrolizumab has been reported to be 2.7% for single agent therapy, and 6.6% for combination therapy [19]. The incidence of pneumonitis is highest in patients with NSCLC and it is typically more severe in these patients [19]. The median time from initiation of therapy to pneumonitis is 2.6 months (range 0.5-11.5 months) [19]. The clinical presentation can be mild to severe [19]. Patterns of pneumonitis include COP (most common), NSIP (next most common), hypersensitivity pneumonitis pattern, and AIP [19]. In all cases, infection should be excluded prior to initiation of therapy [19]. Treatment is to withhold therapy and corticosteroids [19]. Patients that do not response to steroids will require additional immunosuppressant treatment with mycophenolate mofetil, cyclophosphamide, or infliximab (an anti-tumor necrosis factor-alpha immunosuppressant) [19]. Approximately one-third of patients are able to restart immune checkpoint inhibitor therapy after successful treatment for pneumonitis, however, between 25-28% will experience recurrent pneumonitis during retreatment [19]. A pneumonitis flare can also occur during steroid taper prior to restarting therapy [19].
Pembrolizumab is an engineered humanized monoclonal antibody PD-1 inhibitor (thus preventing the inhibition of T-cell activation) that has also been shown to result in improved survival in NSCLC patients [18,25]. The agent is also used for treatment of melanoma and head and neck squamous carcinomas [19]. Immune related adverse events can occur in 29% of patients [18] and include skin reactions, colitis, hepatitis, and pneumonitis [25]. Sarcoid-like reactions (FDG avid mediastinal and hilar adenopathy and skin lesions) have also been reported and can also be seen with other check point inhibitors such as ipilimumab and nivolumab [25]. Interestingly, newer programmed death-ligand 1 (anti PD-L1) inhibitors (durvalumab, avelumab, and atezolizumab) are not thought to be associated with sarcoid-like reactions [25].
Atezolizumab in a anti-PD-L1 monoclonal antibody [18]. The agent is used for treatment of urothelial carcinoma and NSCLC [19].
The most common adverse events associated with PD-1 and PD-L1 inhibitors in patients with lung cancer include skin manifestations (rash and pruritis), hypothyroidism, and pneumonitis [18]. Pneumonitis (which is a diagnosis of exclusion) can be observed in 3-6% of patients treated with PD-1/PD-L1 inhibitors and can be severe and life threatening in 1-3% of cases [18]. The median time to onset of pneumonitis has been reported to be 2.8 months (range 9 days to 19 months) [21]. In patients receiving PD-1 treatment, there is a higher risk for pneumonitis in NSCLC patients compared to melanoma patients [27]. Overall, PD-1 inhibitors have a higher incidence of pneumonitis compared to PD-L1 inhibitors (3.6% vs 1.3%) [27].
The pneumonitis can manifest as COP, NSIP, HP, or AIP, but in one study, COP accounted for approximately 2/3's (65%) of PD-1 inhibitor induced pneumonitis (NSIP 15%, HP 10%, and AIP 10%) [18,21]. The median time for clinical presentation of pneumonitis from initiation of therapy is 15-31 weeks (other authors indicate a median time of 2.6 months [27]) [18]. Symptoms are often non-specific and include new or worsening cough, dyspnea, chest pain, and increasing fatigue [18]. Treatment consists of discontinuance of the offending agent and corticosteroid administration [27]. A subset of cases can be unresponsive to corticosteroids and may require treatment with infliximab (an anti-tumor necrosis factor-alpha immunosuppressant), mycophenolate mofetil, or cyclophosphamide [27]. Up to one-third of patients may be able to restart immune-checkpoint inhibitor therapy after successful treatment of the pneumonitis [27]. However, about 25% of these patients may ultimately experience recurrent pneumonitis ][27]. A small number of patients may experience recurrent pneumonitis after the completion of corticosteroid taper without restarting therapy - this is referred to as a "pneumonitis flare" [27].
CD20 antibody
CD20 is an activated glycosylated phosphoprotein expressed as a surface antigen of B cells during their differentiation that is targeted by the anti-CD20 antibody rituximab (used for the treatment of lymphoma) [19]. Pneumonitis can occur in 4-10% of patients treated with rituximab [19]. Pneumonitis is more common in men, in patients during the 5th and 6th decades of life, and in patients receiving combination therapy with other chemotherapeutic agents [19]. Patients present with diffuse bilateral GGOs and consolidation on chest CT [19]. Treatment is discontinuation of therapy and corticosteroids [19]. Mortality can be as high as 15% [19].
Large focal fat deposits or fatty replacement of intra-abdominal lymph nodes can be seen in patients receiving rituximab therapy, particularly chronic lymphocytic leukemia patients [20].
2- Amiodarone:
Amiodarone is a triiodinated agent used in the treatment of cardiac dysrhythmias that accumulates in the lung largely within macrophages and type II pneumocytes. Pulmonary toxicity is seen in 5 to 10% of patients and usually occurs after 1 to 10 months of therapy. Pulmonary toxicity is dose related and is more likely to occur with doses greater than 400 mg/day. There are two distinct clinical presentations for patients with amiodarone pulmonary toxicity. Most patients present with subacute symptoms of dyspnea and a non-productive cough. A more acute onset mimicking an infectious pneumonitis occurs in about one-third of patients. The most frequent physiologic impairment is a decrease in the carbon monoxide diffusing capacity (DLCO). The prognosis is good and pulmonary toxicity is reversible in the majority of patients after discontinuation of the drug [10]. Clinical symptoms resolve within 2 to 4 weeks and the chest radiographic findings clear slowly over about 3 months.
Radiographic findings may precede the onset of clinical symptoms. Radiographs often demonstrate a diffuse interstitial pattern with patchy alveolar opacities (patients who present with acute symptoms are more likely to have alveolar opacities). CT scan findings include septal thickening, interstitial fibrosis, and high attenuation consolidations due to incorporation of amiodarone into the type II pneumocytes [10]. The infiltrates tend to be peripheral and basilar with attentuation's of 82 to 174 HU). Increased attenuation of the liver and spleen are also characteristic of amiodarone exposure. [3]
3- Gold salts:
Gold salts are used in the treatment of rheumatoid arthritis. The incidence of pulmonary toxicity is less than 1% and is typically a hypersensitivity pneumonitis. Toxicity usually develops 4 to 16 weeks after initiation of therapy, but can occur more rapidly. Patients present with dyspnea, cough, and fever. Eosinophilia is found in about 40% of cases. Reaction is dose related, often treated with steroids, and resolves after the drug is discontinued. Clinical improvement usually occurs over several weeks to months, but patients can have residual pulmonary dysfunction. [3]
The chest radiograph usually shows a basilar reticulonodular pattern, but mixed interstitial-alveolar or purely alveolar opacities have also been described.
4- Nitrofurantoin:
Nitrofurantoin is used to treat urinary tract infections and lung injury is uncommon (under 1% of patients receiving the medication) [6-8]. Because of the frequency with which this medication is prescribed, however, patients presenting with pulmonary toxicity are not uncommon. The agent more commonly produces an acute hypersensitivity pneumonitis usually within 2 weeks of administration with peripheral eosinophilia, fever, dyspnea, and cough [6-8]. Prognosis is good with most patients recovering after discontinuation of the agent [6]. Radiographically, acute toxicity manifests as pulmonary edema with bilateral opacities with a basilar predominance [6,8]. Chronic toxicity is less common and usually occurs after months or years of administration of the agent. Symptoms are incidious in onset and appears identical to chronic interstitial fibrosis.
5- OKT-3:
OKT3 is a murine monclonal antibody directed against the T3 antigen of human T-cells (blocking their normal function) used to treat rejection of allografts. Severe pulmonary edema can complicate the first administration of the agent and is associated with fluid over-load prior to initiation of therapy. [3]
6- Oxygen:
Prolonged therapy with tensions between 80-100% can lead to lung damage.
7- Penicillamine:
Penicillamine is used in the treatment of lead poisoning, WIlson's disease, cystinurea, rheumatoid arthritis, and scleroderma. Pulmonary toxicity is RARE, but four types of toxicity have been described: pulmonary-renal syndrome, bronchiolitis obliterans, chronic alveolitis-interstitial fibrosis, and hypersensitivity pneumonitis.
Bronchiolitis obliterans can occur 3 to 14 months after initiation of treatment. Patients present with subacute onset of cough and dyspnea. Radiographs may be normal, show hyperinflation, or show opacities (reticular or alveolar). Mortality is 38% and residual pulmonary dysfunction persists in those that survive. [3].
Pulmonary-renal syndrome can occur from 10 months to 20 years following initiation of therapy and resembles Goodpasteurs syndrome with alveolar hemorrhage and a necrotizing glomerulonephritis. Affected patients are acutely ill with abrupt onset of dyspnea, cough, hemoptysis, and hematuria. Treatment is with corticosteroids, immunosuppressive agents, and plasmapheresis. Mortality is 36% despite these aggressive measures. [3]
8- Interferon:
Up to one third of patients treated with interferon develop non-specific symptoms of cough, fever, and malaise in the first few months of treatment [9]. Up to 7% of patients on interferon can develop sarcoid [9]. Patients present with fine 1-2 mm nodular opacities which can be identified on both chest radiographs and HRCT [9]. Prognosis for interferon-induced sarcoid is good when the medication can be discontinued [9].
Chimeric antigen receptor (CAR) T-cell therapy:
CAR uses a synthetically created functional cell receptor grafted onto previously harvested patient T cells, which bind to preselected tumor-associated antigens and thereby activate host immune signaling cascades to attack tumor cells [37]. Prior to therapy, the patient undergoes 3 days of conditioning chemotherapy to optimize the environment for CAR T-cells by destroying myeloid suppressor and regulatory T-cells in the circulation [37]. Advantages include a single treatment episode of 2-3 weeks, much more rapid response to therapy than with conventional chemotherapy. and durable disease elimination, with remission rates of over 80% [37]. The treatment is used for recurrent or refractory B-cell lymphomas, B-cell acute lymphoblastic leukemia (ALL), and also multiple myeloma [37]. Agents include tisagenlecleucel, axicabtagene ciloleucel, lisocabtagene maraleucel, brexucabta-gene autoleucel, and idecabtagene vicleucel [37].
Typical imaging response patterns after CAR T-cell therapy demonstrate a decrease in lesion size and a decrease in FDG uptake, usually within one month of therapy [37]. However, up to 40% of patients may not have a complete response to the initial infusion within one month and overall response can be variable for the first three months, and a partial response may improve over time [37]. Psueodprogression can also occur after treatment [37].
Therapy failures include tumor resistance via tumor gene mutation with loss of target antigen and CAR T-cell exhaustion and declining T-cell population [37]. Patients with large pre-therapy disease burden have more severe adverse events and kess durable responses [37].
Complications of CAR therapy:
As the CAR T-cell population expands within the peripheral blood, peaking at approximately day 10 after infusion, simultaneous release of cytokines, including interleukin 6, causes an increased systemic inflammatory response [37]. After 10 day, CAR T-cells increasingly redistribute to the bone marrow and continue to replicate [37]. Immunosuppression with infection risk is increased for the first 3 days after CAR T-cell infusion, primarily from neutropenia due to initial conditioning chemotherapy [37]. Agranulocytosis occurs between days 3 and 10, followed by gradual bone marrow recovery [37]. CAR T-cells can persist in the peripheral blood for over one year and potentially longer [37].
Cytokine release syndrome: CRS is the most common immune therapy toxic effect from the treatment and can be seen in 60-93% of patients [37,38]. CRS affects most patients with ALL and lymphoma that receive CAR T-cell therapy [37]. It predominantly ensues from T-cell activation and stimulation leading to a hyper-immune systemic inflammatory response, including release of cytokines and chemokines, causing marked immune cell aggregation and further increasing levels of inflammatory molecules [37].
Interleukin 6 is known to be a primary driver in CRS and it is secreted primarily by monocytes, macrophages, and dendritic cells [37]. Risk factors for CRS include ALL, large disease burden, high dose of CAR T cells, preexisting thrombocytopenia, and preinfusion lymphodepletion [37]. In severe cases patients may have hypotension/hemodynamic instability, capillary leak, DIC, and hypoxia, which can lead to organ failure [37]. Additional risk factors for severe CRS include rapid onset within 72 hours of infusion [37].
The median time for development of CRS following infusion is 2-3 days, although patients may experience CRS up to 2-3 weeks after infusion [38]. The timing of CRS is also dependent on the therapeutic agent and underlying malignancy [37]. For instance, patients treated with axicabtagene ciloleucel or brexucabta-gene autoleucel, both of which incorporate a CD28 costimulatory domain, have an increased risk for CRS within two days after CAR T-cell administration [37]. For ALL patients treated with tisagenlecleucel, which has a CD137/4-1-BB costimulatory domain, the risk peaks at 3 days [37]. Patients with DLBCL receiving tisagenlecleucel have the highest risk 7 days after infusion [37].
Symptoms of CAS can range from mild, such as fever, to severe with hemodynamic instability and end organ toxicity [38]. Coagulopathy after treatment is reported in up to one-half of patients and acute lung injury can manifest as pneumonitis or ARDS with hypoxia that can lead to mechanical ventilation [37,38]. The presence of pleural effusions and pulmonary edema are considered evidence of respiratory organ toxicity [38]. Major adverse cardiac events can occur in 12-39% of patients [37]. In the abdomen, CT may demonstrate periportal edema, and gallbladder wall thickening [37].
Treatment options involves steroids, supplemental oxygen, hydration, and vasopressors [37]. The first line therapy for severe grade 3 or 4 CRS is tocilizumab, an interleukin 6 blockade agent that has little negative effect on T-cell therapeutic activity [37].
Immune effector cell-associated neurotoxicity syndrome: ICANS is the second most common adverse event after CAR T cell therapy [37]. The prevalence is 23-67% in patients with lymphoma and 40-62% in patients with leukemia [37]. It is often self-limited, but 3% of severe cases can be fatal [37]. ICANS may occur concurrently with CRS, but it may precede or follow it [37]. The onset of ICANS is generally 4 days after CAR T-cell infusion, with a peak at 7 days, but can occur as early as one day after infusion [37]. Resolution is expected by 21-54 days, although a small minority of patients may have longer-term complications [37]. Symptoms include headache and confusion [38].
The mechanism is felt to be related to disruption of the BBB by proinflammatory cytokines, leading to leakage of high levels of cytokines, including interferon-gamma, into the CSF, endothelial activation, and cerebral edema [37]. Risk factors include disease burden and type, prior treatment regimens, volume of CAR T-cell dose, pre-existing neurologic conditions, the preconditioning regimen, and CRS [37]. More serious neurotoxicity has been seen with a CD28 costimulatory domain and is also associated with severe CRS [37].
Characteristic MR findings include symmetric T2-weighted or FLAIR hyperintensities in the periventricular or subcortical white matter and occasional external and extreme capsule involvement [37]. Other authors indicate findings include abnormal T2 signal in the brainstem, thalami, and hippocampus [38].
Corticosteroids are the mainstay of treatment- dexamethasone can be used for grade 2 or 3 ICANS, while high dose IV methylprednisolone is used for grade 4 ICANS [37].
Tumor lysis syndrome: TLS is a serious life-threatening complication caused by rapid tumor breakdown after therapy [37]. Deposition of tumor intracellular components into the circulation causes metabolic abnormalities that can lead to acute kidney injury, arrhythmias, and seizures [37]. It is particularly associated with proliferative tumors such as high-grade NHL and ALL [37]. TLS occurs 0-4 days after therapy initiation [37]. Prophylactic hydration and administration of a hypouricemic agent is advised in patients with large tumor burdens [37].
Infection: Up to 42% of patients contract an infectious process during the first month after treatment, the majority in the first 10 days [37]. About one-third of patients have at least on infection between days 31 and 180 after treatment [37]. Bacterial illness is the most common infection [37]. Viral infection, usually of the respiratory tract, is the second most common infection with a median time of 48 days after treatment [37].
Pegfilgrastim:
Pegfilgrastim is the long acting form of recombinant human granulocyte colony-stimulating factor [45]. Pegfilgrastim therapy can produce an aortitis (symptomatic or asymptomatic- up to 61% of patients) typically within 10 days of administration and most commonly involving the branches of the aortic arch or aortic arch with an incidence of 0.5-2.7% [45,46]. Pegfilgrastim-induced aortitis is more common in older women older than 60 years) [45]. Increased tracer uptake in the aorta can be seen on FDG PET imaging [45]. The condition responds favorably to treatment with steroids [45].
REFERENCES:
(1) Radiol Clin North Am 1991 Sep;29(5):983-997
(2) Radiol Clin North Am 1994 Jul;32(4):731-744
(3) J Thorac Imag 1991; 6 (1): 19-29
(4) J Thorac Imag 1999; Conces DJ. Noninfectious lung disease in immunocompromised patients. 14: 9-24
(5) Clinical Radiology 1992; Padley SP, et al. High-resolution computed tomography of drug-induced lung disease. 46: 232-236
(6) Society of Thoracic Radiology Annual Meeting 2000 Course Syllabus; Erasmus JJ. Pulmonary drug toxicity: Pathogenesis and radiologic manifestations. 65-68
(7) Radiographics 2000; Rossi SE, et al. Pulmonary drug toxicity: Radiologic and pathologic manifestations. 20: 1245-1259
(8) AJR 2000; Ellis SJ, et al. Drug-induced lung disease: High-resolution CT findings. 175: 1019-1024 (No abstract available)
(9) AJR 2001; Ravenel JG, et al. Sarcoidosis induced by interferon therapy. 177: 199-201
(10) AJR 2005; Marchiori E, et al. Diffuse high-attenuation pulmonary abnormalities: a pattern-oriented diagnostic approach on high-resolution CT. 184: 273-282
(11) Radiol Clin N Am 2005; Lindell RM, Hartman TE. Chest imaging in iatrogenic respiratory disease. 43: 601-610
(12) Radiology 2011; Tossisi JM, et al. CT findings of chemotherapy induced toxicity: what radiologists need to know about the clinical and radiologic manifestations of chemotherapy toxicity. 258: 41-56
(13) AJR 2013; Abramson RG, et al. COmplications of targeted drug therapies for solid malignancies: manifestations and mechanisms. 200: 475-483
(14) Radiographics 2014; Viswanathan C, et al. Abdominal and pelvic complications of nonoperative oncologic therapy. 34: 941-961
(15) Radiographics 2014; Choi MH, et al. Acute pulmonary complictions in patients with hematologic malignancies. 34: 1755-1768
(16) AJR 2014; Rohatgi S, et al. Vascular toxicity associated with chemotherapy and molecular targeted therapy: what should a radiologist know? 203: 1353-1362
(17) Radiographics 2015; Tirumani SH, et al. Anti-VEGF molecular targeted therapies in common solid malignancies: comprehensive update for radiologists. 35: 455-474
(18) AJR 2017; Wang GX, et al. Immune checkpoint inhibitors in lung cancer: imaging considerations. 209: 567-575
(19) Radiographics 2017; Nishino M, et al. Thoracic complications of precision cancer therapies: a practical guide for radiologists in the new era of cancer care. 37: 1371-1387
(20) Radiographics 2017; Chang ST, et al. Molecular and clinical approach to intra-abdominal adverse effects of targeted cancer therapies. 37: 1461-1482
(21) Radiographics 2017; Wang GX, et al. Immune checkpoint inhibitor cancer therapy: spectrum of imaging findings. 37: 2132-2144
(22) J Nucl Med 2017; Gonzalez Trotter DE, et al. In vivo imaging of the programmed death ligand 1 by 18F PET. 58: 1852-1857
(23) J Nucl Med 2018; Donnelly DJ, et al. Synthesis and biologic evaluation of a novel 18F-labeled adnectin as a PET radioligand for imaging PD-L1 expression. 59: 529-535
(24) AJR 2018; O'Neill AC, et al. Hallmarks of cancer in the reading room: a guide for radiologists. 211: 470-484
(25) Radiology 2018; Cheshire SC, et al. Pembrolizumab-induced sarcoid-like reactions during treatment of metastatic melanoma. 289: 564-567
(26) Radiology 2018; Nobashi T, Mittra E. PD-1 blockade-induced inflammatory arthritis. 289: 616
(27) Radiology 2019; Nishino M, et al. Imaging of cancer immunotherapy: current approaches and future directions. 290: 9-22
(28) Radiographics 2019; Kalisz KR, et al. Immune checkpoint inhibitor therapy-related pneumonitis: patterns and management. 39: 1923-1937
(29) J Nucl Med 2020; Castello A, et al. Hyperprogressive disease in patients with non-small cell lung cancer treated with checkpoint inhibitors: the role of 18F-FDG PET/CT. 61: 821-826
(30) J Nucl Med 2020; Iravani A, Hicks RJ. Imaging the cancer immune environment and its response to pharnacologic intervention, part 1: the role of 18F-FDG PET/CT. 61: 943-950
(31) J Nucl Med 2020; Lutje S, et al. Immune checkpoint imaging in oncology: a game changer toward personalized immunotherapy? 61: 1137-1144
(32) AJR 2020; Thomas R, et al. A review of the mechanisms and clinical implications of precision cancer therapy-related toxicity: a primer for the radiologist. 215: 770-780
(33) Radiology 2020; Park HJ, et al. Incidence of pseudoprogression during immune checkpoint inhibitor therapy for solid tumors: a systematic review and meta-analysis. 297: 87-96
(34) Radiorgaphics 2020; Garcia-Figueiras R, et al. Assessing immunotherapy with functional and molecular imaging and radiomics. 40: 1987-2010
(35) Radiology 2021; Johkoh T, et al. Chest CT diagnosis and clinical management of drug-related pneumonitis in patients receiving molecular targeting agents and immune checkpoint inhibitors: a position paper from the Fleishner society. 298: 550-566
(36) Radiology 2021; Faron A, et al. Cardiac MRI depicts immune checkpoint inhibitor induced myocarditis. a prospective study. 301: 602-609
(37) Radiographics 2022; de Groot PM, et al. Imaging primer on chimeric antigen receptor T-cell therapy for radiologists. 42: 176-194
(38) Radiology 2022; Smith DA, et al. Imaging-based toxicity and response pattern assessment following CAR T-cell therapy. 302: 438-445
(39) J Nucl Med 2022; Westdorp H, et al. Toward a better understanding of immune checkpoint inhibitor radiolabeled PET imaging studies. 63: 359-361
(40) Radiology 2022; Cadour F, et al. Cardiac MR features and prognostic value in immune checkpoint inhibitor-induced myocarditis. 303: 512-521
(41) AJR 2022; Sheikhbahaei S, et al. Imaging of cancer immunotherapy: response assessment methods, atypical response patterns, and immune-related adverse events, from the AJR special series on imaging of inflammation. 218: 940-953
(42) Radiology 2022; Hughes DJ, et al. 18F FDG PET/CT and novel molecular imaging for directing immunotherapy in cancer. 304: 246-264
(43) Radiographics 2022; Hata A, et al. Interstitial lung abnormalities at CT: subtypes, clinical significance, and associations with lung cancer. 42: 1925-1939
(44) Radiographics 2022; Shroff GS, et al. Imaging of immune checkpoint inhibitor immunotherapy for non-small cell lung cancer. 42: 1956-1974
(45) Radiology 2022; Takamatsu A, et al. Single-center analysis of pegfilgrastim-induced aortitis using a drug prescription database and CT findings. 305: 729-740
(46) Radiology 2022; Krinsky G. Pegfilgrastim-induced aortitis: cardiovascular diagnosis in the oncologic setting. 305: 741-742
(47) Radiology 2023; Dercle L, et al. Emerging and evolving concepts in cancer immunotherapy imaging. 306: 32-46
(49) AJR 2023; Murphy DJ, et al. Imaging follow-up of nonsurgical therapies for lung cancer: AJR expert panel narrative review. 221: 409-424om the Fleishner society. 298:
550-566