Top Story

Latest News

ChatGPT performs poorly on ACR exam for residents
April 24, 2024

Sponsored
Register today for a FREE webinar May 8 at 12 Noon EDT
April 24, 2024

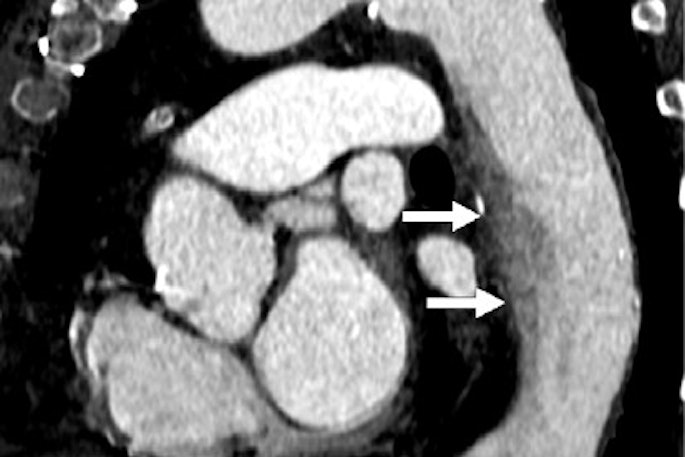
CT reveals influence of diabetes on COVID-19 severity
April 25, 2024

State of the RT profession more palpable than ever
April 24, 2024
Cases of the Week
Check out our Cases of the Week!





More from AuntMinnie
RaySearch Laboratories publishes full 2023 report
April 26, 2024

USPSTF solicits comment on draft recs for CVD assessment
April 26, 2024

Fujifilm launches septal myectomy aid
April 26, 2024

New Neiman study shows impact of nonphysician providers
April 26, 2024

Advanced visualization in 2024: Enhancing radiology care
April 25, 2024

Median posts Q1 results, reports net loss for 2023
April 25, 2024


ScreenPoint CEO steps down; new CEO named
April 25, 2024


Accuray to showcase radiation therapy tech at ESTRO
April 25, 2024

Provisio's SLT IVUS System gets FDA nod
April 25, 2024

