Immunodeficiency Syndromes:
Thoracic abnormalities are present in up to 60% of patients with primary humeral immunodeficiency [6].
B-cell Deficiency:
T-cell Deficiency:
Combined T and B-cell Deficiency:
Phagocytic Immundeficiency:
B-cell Antibody Deficiencies:
Congenital X-linked Hypogammaglobulinemia/ Agammaglobulinemia (Bruton's agammaglobulinemia):
An autosomal recessive abnormality which is seen only in males (x-linked recessive disorder). This disorder is characterized by disruption of all B-cell development phases [6]. Affected patients have markedly decreased numbers of mature B-cells (less than 1%) and plasma cells in the circulation and consequently they have reduced amounts of lymphoid tissue [2,6]. There is severely decreased or absent levels of all immunoglobulins [6]. T-cell mediated immunity remains intact. During the first 6-9 months of life, these patients are protected from infections by circulating maternal IgG antibodies [2]. As these levels decrease, patients develop recurrent pyogenic sino-pulmonary infections which lead to bronchiectasis. About 20% of cases do not present in infancy (between ages 3-5 years)- probably due to the widespread use of antibiotics [2]. Hypoplasia of the adenoids, tonsils, and peripheral lymph nodes is seen, while the thymus appears normal. Splenomegaly is not seen [2]. Treatment consists of IV immunoglobulin [2]. These patients have an increased risk of leukemia/lymphoma (seen in about 6% of patients).Common Variable Immunodeficiency:
A progressive deficiency in cell mediated immunity with no (possible?) genetic transmission that presents in late childhood/early adulthood it is characterized by recurrent sino-pulmonary infections which leads to bronchiectasis. Antibody production of all major classes is impaired [6]. In this disorder, B-cells are normal in number and phenotype, but are unable to produce immunoglobulins or are unable to secret immunoglobulins once they are produced. T-cell-mediated immunity is often intact, but T-cell abnormalities may be present in up to 60% of patients [2]. Patients have a healthy amount of tonsillar tissue and lymphadenopathy or splenomegaly is found in 15-25% of patients [2]. There is no gender prevalence [6]. About 20% of affected patients develop autoimmune diseases [2].Treatment consists of IV immunoglobulin replacement [2]. There is a 8-10% incidence of lymphoma (high grade B-cell lymphoma)/leukemia, as well as an increased incidence of gastric cancer and thymoma (the presence of a thymoma in association with immunodeficiency is known as Good syndrome [6]. The prevalence of lymphoma is 30 times higher than that in the general population [6].
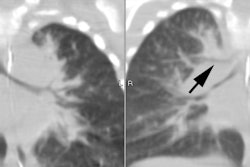
Granulomatous and lymphocytic interstitial lung disease is a distinct ILD that occurs in patients with common variable immune deficiency [7]. The overall prevalence of interstitial lung disease in patients with combined variable immunodeficiency was 26%, with two-thirds of these patients demonstrating GL-ILD on biopsy [7]. The most common imaging finding in GL-ILD is diffuse lower lobe predominant micronodules [7]. Most cases also demonstrate interlobular septal thickening [7].
T-cell (Cellular) Deficiencies:
DiGeorge's Syndrome:
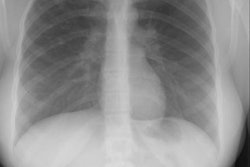
A congenital immunodeficiency due to gene defects on chromosome 22 which leads to failure of development of the 3rd and 4th pharyngeal pouches [2]. There is an absent/hypoplastic thymus (that produces a defect in cell mediated immunity) and absent parathyroid glands. Cardiac anomalies can also occur [2]. Patients also have dysmorphic facies with micrognathia, low set ears, shortened philtrum of the upper lip, celft palate, hypertelorism, congenital heart disease (including truncus arteriosus, interrupted aortic arch [about 10% of patients], tetrology of Fallot, and absent pulmonary valve), and neonatal hypocalcemic tetany (due to hypoparathyroidism).B-cells are present in normal numbers, but antibody response may abnormal because of an inadequate number of T-cells (which varies depending on the degree of thymic hypoplasia) [2]. Patients are at an increased risk for opportunistic infections such as viral, fungal, acid-fast bacteria, and PCP infections. There is no increased risk of malignancy. In up to 80% of patients, the immunodeficiency is mild (or partial DiGeorge's) and can even be transient [2]. Treatment is usually supportive [2]. Thymic epithelial transplants or unfractionated human leukocyte antigen-identical sibling bone marrow transplantation is recommended only for patients with complete DiGeorge's syndrome [2]. The CXR will reveal a narrowed superior mediastinum and retrosternal lucency due to absence of the thymus.
Combined T and B-cell Immunodeficiency:
Severe Combined Immunodeficiency Disease:
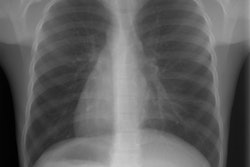
There is absence of both cellular (T-cell) and humoral (B-cell) immunity. The disorder is inherited by either an autosomal recessive or X-linked pattern (46% of cases) [2]. Patients present within the first months of life with frequent episodes of pneumonia, otitis, sepsis, persistent oral thrush, and cutaneous infections. Immunization with attenuated viruses should be avoided as this could prove fatal [2]. Without immune reconstitution patients rarely survive beyond 1 year of age. Bone marrow transplant is presently the preferred therapy [2]. On CXR these patients also have an absent thymic shadow [2].Ataxia telangiectasia (:ouis-Barr's syndrome):
Ataxia telangiectasia is an autosomal recessive combined immunodeficiency disorder seen in early childhood with a combination of cutaneous and scleral telangiectasias, progressive cerebellar ataxia, and recurrent sino-pulmonary infections. Immunologic features include selective IgA deficiency or hypogammaglobulinemia and T-cell dysfunction is moderately severe [2]. The thymus and lymphoid tissue are hypoplastic and there is a 10% increased risk for lymphoreticular malignancies. Treatment is limited to supportive care and no cure is available [2].Wiskott-Aldrich Syndrome (Partial combined immunodeficiency syndrome):
This is a rare X-linked recessive disease characterized by eczema, thrombocytopenia with small defective platelets, and recurrent pyogenic infections usually before 1 year of age. The gene on the X-chromosome responsible for the condition encodes a protein called the Wiskott-Aldrich protein and is expressed in lymphocytes, the spleen, and thymus [2]. The protein is thought to play a role in actin polymerization [2]. Immunologically, there is impaired antibody responses to polysaccharide-encapsulated organisms (such as pneumococci, H. influenzae, and meningococci) [2]. Affected patients have a characteristic serum immunoglobulin pattern of elevated IgA and IgE, normal or slightly decreased IgG levels, and decreased IgM levels [2]. Affected infants typically first present with bleeding abnormalities and pyogenic infections begin to develop during the first year of life [2]. There is an increased risk (8%) for lymphoreticular malignancies. Patients rarely survive beyond their teenage years without bone marrow transplant [2].Phagocytic Immunodeficiency:
Chronic Granulomatous Disease:
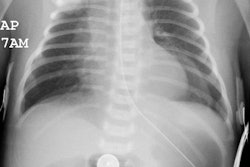
CGD results from a deficiency in production of hydrogen peroxidase and oxygen radicals in macrophages and polymorphonuclear cells. This defect is characterized by leukocyte dysfunction (infectious organisms are phagocytized normally, but are not killed) that results in an increased susceptibility to bacterial and fungal infection particularly with catalase-positive organisms [4]. The disorder is most commonly (two-thirds of patients) an X-linked recessive disorder (affecting males-86% of cases [5]), but the disorder can also occur in females due to an autosomal recessive defect in the nicotinamide adenine dinucleotide phosphate (NAPDH) oxidase system. S. aureus and Aspergillus organisms are the most common causes of infection [1]. These organisms are catalase- positive and therefore destroy any oxygen radicals that they produce [2]. Prolonged intracellular existence of these catalase-positive microorganisms triggers a cell-mediated response that results in granuloma formation [2]. Patients usually present as within the first few months of life with pneumonia (the most common site of infection-typically fungal [2]), adenopathy/suppurative adenitis, hepatosplenomegally (up to 90% of patients [4]), and osteomyelitis (common sites include the ribs, vertebrae, and small bones of the hands and feet [4]). Other findings include gastric antral narrowing which occurs in up to 16% of patients [4]. In approximately 10% of patients, diagnosis is not made until the second decade of life [5]. Currently approximately 50% of patients with CGD survive through the third or fourth decade of life [5].Chediack-Hoigashi Syndrome:
This is also a rare autosomal recessive disorder caused by impaired chemotaxis and bacterial-killing functions [2]. Most patients do not survive beyond age 10 years. The disorder is characterized by granulocyte dysfunction, recurrent bacterial infections, partial oculocutaneous albanism, other eye problems, peripheral neuropathy, and an increased incidence of malignancy. Affected children are susceptible to both bacterial and Ebstein-Barr infections. Treatment of choice is bone marrow transplantation [2].REFERENCES:
(1) J Thorac Imaging 1999; Pennington DJ, et
al. Pulmonary disease
in the immunecompromised child. 14: 37-50
(2) AJR 2001; Yin EZ, et al. Primary immunodeficiency disorders in pediatric patients: Clinical features and imaging findings. 176: 1541-1552
(3) J Nucl Cardiol 2003; Grossfeld PD. The genetics of congenital heart disease. 10: 71-76
(4) Radiographics 2005; Khanna G, et al. Imaging chronic granulomatous disease in children. 25: 1183-1195
(5) AJR 2008; Godoy MCB, et al. Chest radiographic and CT manifestations of chronic granulomatous disease in adults. 191: 1570-1575
(6) Radiographics 2009; Bierry G, et al. Thoracic manifestations
of primary humoral immunodeficiency: a comprehensive review. 29:
1909-1920
(7) Radiographics 2020; Naeem M, et al. Noninfectious granulomatous diseases of the chest. 40: 1003-1019