Cystic Fibrosis:
View cases of cystic fibrosis
Clinical:
Cystic fibrosis (CF) is an autosomal recessive disorder affecting the long arm of chromosome 7 which encodes a large single-chain protein that forms a chloride channel and its associated regulatory regions (CF transmembrane conductance regulator). Over 70% of patients with CF have a deletion of three contiguous base pairs resulting in the loss of a single amino acid in the chain (deletion of phenylalanine at codon 508) [4]- referred to as the ΔF508 mutation [10]. This deletion results in a misfolded CFTR protein that prevents trafficking into the cells apical membrane [10].CF is characterized by thickened bronchial mucus secretions which cannot be cleared due to reduced mucociliary transport. The initial histologic lesion in CF patients is bronchiolitis which leads to obstruction of the small airways and air trapping (emphysematous changes). Symptoms include recurrent pneumonia's, pansinusitis and nasal polyps, infertility in males, and an increased sodium chloride concentration in sweat (greater than 60 mEq/L). Non-classic cases can be seen and up to 7% of CF cases are not diagnosed until after the age of 16 years [10]. These patients often have a so-called "mild" CF mutation and have some retained chloride channel activity [10].
The disease is most often found in whites (95%), and is rare in blacks. Males and females are affected with nearly equal frequency. About 10% of cases will escape detection until the patient is in late adolescence or early adulthood. Recurrent infections, initially with Staphylococcus aureus and Pseudomonas aeruginosa are common due to the tenacious obstructing secretions and eventually lead to bronchiectasis. Staphylococcus aureus infection and colonization is now more effectively controlled with the use of antistaphylococcal antibiotics [4]. Pseudomonas aeruginosa is now more prevalent, causing chronic infection in up to 90% of adults [4]. Other organisms such as aspergillus (10-20% of CF patients), nontuberculous mycobacteria, Burkholderia cepacia, and Pseudomonas cepacia are becoming increasingly important pulmonary pathogens in older patients with CF [4,10].
Chronic inflammation also leads to fibrosis. Other complications include pneumothorax (5 to 20% of patients due to subpleural bleb rupture [4]), hemoptysis (up to 60% of patients at some time [4]), hypertrophic pulmonary osteoarthropathy (associated with increasing age and disease severity [4], pulmonary arterial hypertension, and cor pulmonale. Cor pulmonale and right heart failure is a poor prognostic indicator in adults, associated with a mean survival of 8 months [4].
As a result of aggressive therapy, more patients with CF are living into their 20's and 30's. Cystic fibrosis is no longer just a pediatric disease as the median survival is presently 29 years [4] and individuals born since the year 2000 will have a median life expectancy of 50 years [9]. Currently, 40-50% of CF patients are adults [10]. Double lung transplantation has been used to treat a limited number of cystic fibrosis patients. The pulmonary secretory defect has not recurred in transplanted lungs [1].
Extrapulmonary manifestations are also common. Nearly 100% of
patients with CF develop chronic rhinosinusitis - the most common
pathogens are S. aureus and P. aeruginosa [10].
Interestingly, frontal sinus hypoplasia has been reproted in
44-66% of adult CF patients [10].
Between 85-90% of adult patients with CF diagnosed in childhood have deficient pancreatic enzyme production and pancreatic exocrine insufficiency (found in up to 90% of CF patients by age 9 years), which leads to fat malabsorption, steatorrhea, malnutrition, and a deficiency of fat-soluble vitamins (including vitamin D) [10]. Complete fatty replacement of the pancreas is the most common pancreatic abnormality seen on CT (56-93% of patients) [6]. Small 1-3 mm pancreatic cysts are commonly seen [6]. Pancreatitis or pancreatic inflammation may occur in association with pancreatic insufficiency, but this is not common [7]. Diabetes (endocrine dysfunction) develops in 1% of children, and 15-50% of adults with CF and pancreatic insufficiency [5,6].
Hepatic involvement is found in 20-50% of patients can results in steatosis, hepatomegaly, and focal biliary cirrhosis [6]. Frank cirrhosis is uncommon (less than 5% of patients) [6]. A microgallbladder is found in 25% of patients at autopsy [6].
Generally, all infants presenting with meconium ileus are later diagnosed with CF [6]. Meconium ileus is found at birth in 10-20% of cases, and subsequent episodes of bowel obstruction (meconium ileus equivalent or distal intestinal obstruction syndrome) occur in 20-25% of cases- possibly related to thickened intestinal mucus. The frequency of episodes of bowel obstruction increase with age. Patients typically present with crampy abdominal pain and a palpable low abdominal mass. CT findings include colonic wall thickening, mural striations, mesenteric soft tissue infiltration, and increased pericolic fat [6]. Treatment for these episodes involves balanced electrolyte fluid lavage and initiation of pancreatic enzyme treatment. Colonic strictures (fibrosing colonopathy) may also be a cause of distal bowel obstruction and is most commonly seen in children younger than 10 years of age [6]. The strictures often involve long segments of the colon and are associated with high dose pancreatic enzyme replacement [6]. The appendix is routinely enlarged (over 6 mm) due to mucoid impaction [6].
Distal ileal obstructive syndrome (DIOS- meconium ileus
equivalent in adults) is a relatively common complication that
occurs in 10-15% of CF patients [8]. It is thought to be related
to a combination of accumulation of thickened intralumenal
secretions and undigested food in the distal small bowel and
cecum, and delayed transit [8,10]. Patients present with
acute-onset periumbilical or RLQ pain with nausea and vomiting
[8]. Risk factors for DIOS include pancreatic insufficiency,
history of meconium ileus or DIOS, and dehydration [8]. The
initial approach to treatment is empirical, with most patients
responding to oral rehydration and osmotic laxatives [10].
Ileocolic intussusception is a known complication of CF seen more
commonly in older children (average age of 10 years) [6]-
incidence of 1% [8]. The lead mass is thought to be due to
inspissated stool [10].
Congenital bilateral absence of the vas deferens is almost
universal in male subjects with classic CF [10]. Although up to
98% of men with CF are azospermic, spermatogenesis is normal and
viable sperm can be harvested microscopically from epididymal
remnants [10].
X-ray:
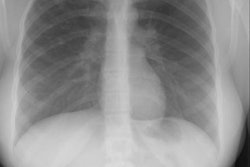
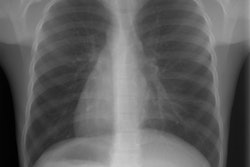
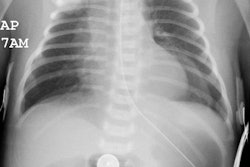
CXR: On plain radiograph there is hyperaeration with prominent lung markings. Hyperaeration is the earliest sign of CF and results from mucous plugging of small bronchioles [4]. Early in the disease involvement is predominantly in an upper lobe distribution, especially the right upper lobe [4]. As the disease progresses, all lobes become involved. Underlying bronchitis results in peribronchial cuffing and bronchial wall thickening. Both the central and peripheral bronchi are abnormal in a majority of patients. Thickening of the wall of the right upper lobe bronchus (best seen on the lateral view), can be an early sign of disease. Cylindrical bronchiectasis produces a "tram track" appearance. Mucoid impaction is very common (up to 70% of patients) and produces typical "finger-like" mucous plugs and subsegmental atelectasis. A clue to the diagnosis is also provided by paranasal sinus films as these patients invariable also suffer from pansinusitis. Although pneumothorax occurs in 5 to 20% of patients, large pneumothoraces are uncommon. This is because patients with CF tend to have very non-compliant lungs which do not easily collapse- as a result, even a small amount of pleural area may produce a tension pneumothorax. Hilar lymph node enlargement is common due to the chronic pulmonary infections [4]. Pleural effusions are not common in CF despite the chronic infections [4].Computed tomography: HRCT can show morphologic abnormalities in asymptomatic cystic fibrosis patients with normal CXR's [3]. Findings in patients with cystic fibrosis include cylindrical bronchiectasis or "signet ring" sign which is found in all patients with advanced cystic fibrosis. Bronchial wall thickening is also commonly present. Areas of consolidation due to atelectasis or super-imposed infection can be seen. In the lung periphery, mucoid impaction of the small bronchioles and associated peribronchiolar inflammation will produce nodular or "tree-in-bud" centrilobular opacities. Geographic areas of decreased lung opacity reflect the presence of air trapping and produce a pattern of mosaic attenuation. Small areas of air trapping can coalesce to form large cysts. Large blebs may form in the apices and can rupture resulting in a pneumothorax. End-stage lung changes with pulmonary hypertension and cor pulmonary are often the cause of mortality in cystic fibrosis patients.
REFERENCES:
(1) Radiology 1997; Cystic fibrosis: 1997. 204(1), 1-10 (no abstract available)
(2) Radiol Clin North Am, May 93, p.617-630
(4) Radiol Clin North Am 1998; Ruzal-Shapiro C. Cystic fibrosis: An overview. 36 (1): 143-161
(5) Radiology 1999; Soyer P, et al. Cystic fibrosis in adolescents and adults: Fatty replacement of the pancreas- CT evaluation and functional correlation. 210: 611-615
(6) AJR 2006; Fields TM, et al. Abdominal manifestations of cystic fibrosis in older children and adults. 187: 1199-1203
(7) AJR 2007; Haber HP. Cystic fibrosis in children and young
adults: findings on routine abdominal sonography. 189: 89-99
(8) Radiographics 2015; mLavelle LP, et al. Cystic fibrosis below
the diaphragm: abdominal findings in adult patients. 35: 680-695
(9) AJR 2016; Murphy KP, et al. Imaging of cystic fibrosis and
pediatric bronchiectasis. 206: 448-454
(10) AJR 2017; Averill S, et al. Multisystem imaging findings of
cystic fibrosis in adults: recognzing typical and atypical
patterns of disease. 209: 3-18