Idiopathic Pulmonary Hemosiderosis:
View cases of idiopathic pulmonary hemosiderosis
Clinical:
A rare probably autoimmune process which results in widespread hemosiderin deposition in the lungs. The disorder is seen most commonly in children under the age of 10 years (typically between the ages of 1 to 7 years) and affects males and females equally. In adults, males are affected more than females and the disease is usually less severe than in children. Patients present with recurrent hemoptysis (acute attacks which last 2-4 days and is usually not massive), pulmonary infiltrates due to alveolar hemorrhage, iron deficiency anemia, weakness, clubbing, and hepatosplenomegaly (25%). Prognosis is poor with recurrent episodes and remissions. Patients generally die within 5 years due to cor pulmonale. The disorder is occasionally associated with celiac disease- see below, or immunoglobulin A gammopathy.Lane-Hamilton syndrome is idiopathic pulmonary hemosiderosis in association with celiac disease [4]. This is most commonly seen in children younger than 15 years of age (although cases have been reported in adults) [4]. The treatment of the celiac disease with a gluten-free diet can lead to remission of the idiopathic pulmonary hemosiderosis in these patients [3,4].
X-ray:
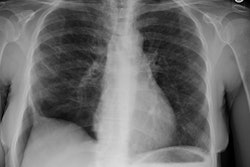
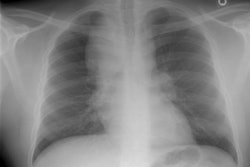
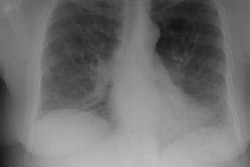
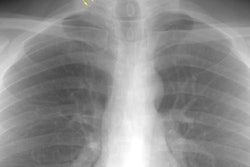
CXR: Following acute hemorrhage there are patchy parenchymal infiltrates with a perihilar and lower lung predominance that may resemble a pneumonia. Bilateral, hazy parahilar opacities can be seen and may mimic pulmonary edema. The infiltrates typically clear over 5 to 7 days. A reticulonodular interstitial pattern develops as blood is cleared from the alveoli and hemosiderin is deposited within the septa. Later there is interstitial fibrosis. Adenopathy and pleural effusions are infrequent findings.HRCT: There are large areas of lobular of diffuse ground glass attenuation representative of areas of hemorrhage. Smooth interlobular septal thickening is present in most cases [3].
REFERENCES:
(1) AJR 1995 Feb;164(2):295-300
(2) Radiol Clin North Am 1991 Sep;29(5):965-971
(3) Radiographics 2002; Engelke C, et al. High-resolution CT and CT angiography of peripheral pulmonary disorders. 22: 739-764
(4) Radiology 2010; Nishino M, et al. Lane-Hamilton syndrome. 254: 985-988