Eosinophilic Granuloma:
(Langerhans Cell Granulomatosis or Langerhans Cell Histiocytosis)
View Images of eosinophilic granuloma
Langerhans' cell histiocytosis (Histiocytosis X) describes a group of syndromes who share a common pathologic feature of infiltration of involved tissues by Langerhans' cells [12]. Classically, there is involvement of the skeletal system with a characteristic lytic bone lesion (young children) or a more acute disseminated form which occurs in infants. Pulmonary involvement is not unusual in systemic forms of Langerhans' cell histiocytosis (LCH), but symptoms are rarely prominant [12]. Localized pulmonary Langerhans'cell histiocytosis (also sometimes referred to as pulmonary eosinophoic granuloma) is a rare pulmonary disease that occurs predominantly in young adults. It has several unique clinical features which justify its classifcation as a distinct clinicopathologic entity [12]. The precise incidence and prevalence of pulmonary LCH are unknown, although studies of lung-biopsy specimens from patients with interstitial lung disease identified pulmonary LCH in only 5% of cases [13].
Clinical:
Pulmonary involvement in LCH is characterized by a granulomatous infiltration of the alveolar septae and bronchial walls with Langerhans' cells which produce small nodules (1 mm to 5 mm in size). The infiltrate can lead to progressive lung destruction and widespread cystic change. The disorder is rare, accounting for only 3.4-5% of cases of chronic diffuse interstitial lung disease. Patients with isolated pulmonary involvement are usually young adults (age 20 to 40 years), Caucasian, and there is an equal frequency in men and women (although young men appear to have the worst prognosis). The disorder is rare in blacks. There is a strong association with smoking (90-95% of cases occur in smokers) and symptoms often improve with cessation of smoking. There are no known genetic factors that predispose persons to pulmonary LCH [13]. Between one-quarter and one-third of patients are asymptomatic at presentation. Symptomatic patients may present with cough (66%), progressive dyspnea, weight loss, fever, or diabetes insipidus (25%). Pneumothorax occurs in 10-25% of patients and will recur on the ipsilateral side in up to 58% of patients [16]. Hemoptysis occurs in less than 5% of patients [13]. There is no consistent pulmonary function test finding, but mild airway obstruction is common. A reduction in the carbon monoxide diffusing capacity is the more common PFT abnormality and is present in 60% to 90% of cases [13]. On bronchoalveolar lavage (BAL), there is an increased number of cells, particularly macrophages, but this may merely be a manifestation of patients smoking history [12]. Langerhans' cells can also be found on BAL, but this finding is not specific for the disorder [12,13]. When the proportion of Langerhans' cells in the BAL fluid is greater than 5%, a diagnosis of pulmonary LCH is very likely [13]. However, a confident diagnosis of pulmonary LCH can often be made based upon the patients age, smoking history, and characteristic HRCT findings- especially if patients are to be followed without treatment [12]. When tissue confirmation is required, the diagnosis is best make by open lung biopsy via video-assisted thoracoscopy [12,13]. Transbronchial biopsy has a low diagnostic yield (ranging from 10-40%) because of the patchy nature of the disease and the small amounts of tissue obtained [12,13].
The course of pulmonary disease is variable- remission occurs in about 25-30% of patients, stabilization in 30-50%, and progression in 25-30% [2,9,17]. Death from respiratory failure or cor pulmonale occurs in 5% [9]. Asymptomatic or minimally symptomatic patients tend to have the best outcomes [13]. Decreased survival is associated with an very young or old age at diagnosis, persistence of systemic symptoms, recurrent pneumothoraces, extrathoracic disease (except bone lesions), diffuse cystic lesions on CXR, a lower FEV1/FVC ratio at diagnosis, a higher RV/TLV ratio at diagnosis, and need for steroid therapy [7,12].
Treatment consists of cessation of smoking which leads to stabilization of symptoms in most patients [13]. Corticosteroids are used for progressive or systemic disease [13]. Cytotoxic agents (such as cyclophosphamide) can be employed for patients that do not respond to smoking cessation and steroids [13]. No treatment has been confirmed to be useful and no double-blind therapeutic trials have been reported [12,13]. Lung transplantation has also been performed for treatment of LCH. There are no specific guidelines for which patients with pulmonary LCH should be candidates for lung transplantation [13]. Patients with rapidly declining lung function, severe pulmonary symptoms, and lack of response to other treatments are the best candidates for lung transplant consideration [13], but the disorder may recur in the transplanted lung in up to 25% of patients [8,9,15]. Pleurodesis may be required for effective therapy of recurrent pneumothorax, however, this may complicate subsequent lung transplant surgery for patients with progressive disease [16].
The etiology of the disorder is not known, but the bronchocentricity and tendency to regress following cessation of smoking suggest a reactive immune response in the bronchioles to an inhaled antigen in cigarette smoke mediated by the Langerhans' cell system [4,8,13]. The recurrence of the disease after transplantation suggests that extrapulmonary factors also play a role in disease pathogenesis, or that the condition may represent a neoplastic disorder [8]. An association between EG and lymphoma has been described [12,13]. Bronchogenic carcinoma has been identified with increased frequency in patients with pulmonary LCH [12]. Lung scaring and smoking may contribute to the development of lung cancer in these cases [12,13]. Although intriguing, there is likely insufficient evidence either to prove or to refute the association between pulmonary LCH and malignanct neoplasms [13].
Histopathology:
Langerhans' cells are part of the widespread system of "dendritic cells" which arise from bone marrow stem cells and they are normally present in the human lung [7-9]. They can be distinguished from histiocytes by cytoplasmic immunostaining with S100 antigen [8,9]. Two other unique features of Langerhans' cells are the presence of Birbeck granules (rod-shaped intracellular structures identified by electron microscopy) and the presence of CD1a antigen on the cell surface [13]. As a result, some authors prefer to term the disorder Langerhans cell "granulomatosis" as the Langerhans cell is not a member of the mononuclear phagocytic system (ie: not a macrophage or "histiocyte") [7]. In LCH, there is an abnormal non-malignant proliferation of monoclonal Langerhans cells [18]. Because Langerhans' cells may be identified in several pathologic pulmonary processes, the mere presence of pulmonary Langerhans' cells is not diagnostic of pulmonary LCH [13].
X-ray:
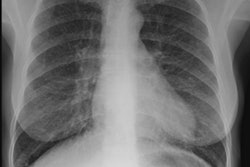
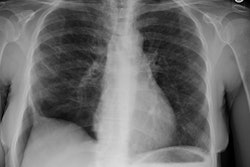
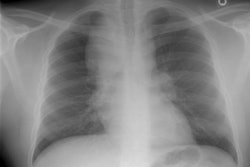
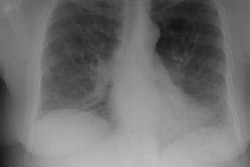
CXR: The CXR is normal in under 10% of cases [12].Early in the disease there is usually diffuse reticulonodularity (3-10 mm nodules which may cavitate) in a symmetric upper lobe predominance (bases tend to be spared) with preservation of lung volumes [13,17]. The nodules demonstrate irregular borders [17]. Associated pneumothorax is found in 15-25% of cases. Pleural effusion is rare. In the late stage, there may be diffuse cysts which spare only the costophrenic angles.
Based upon the radiographic findings, disorders which can be entertained in the differential diagnosis for LCH include: Sarcoid, Silicosis, hypersensitivity pneumonitis, and lymphangioleiomyomatosis (during the later stages of the disorder when cytic lesions predominate) [13].
Computed tomography: On HRCT the distribution of the disease is similar to that on CXR with an upper lobe predominance (likely related to the inhalational component of the disorder [18]). In the early stages, findings include centrilobular opacities and small nodules (1 to 5 mm, but up to 1.5 cm) [17]. The nodules may be few or innumerable and typically have irregular margins [17]. Later in the disorder there is cystic cavitation of small nodules, and cysts- initially thick walled and later thin walled (typically lesions progress in this fashion) [17]. Some authors feel that the nodules do not cavitate and that the cysts represent paracicatrical emphysematous change adjacent to the nodules. The cysts are usually less than 10 mm in size, although cysts larger than 10 mm are found in over half the cases. The walls of the cysts are usually thin (1mm or less), but can be variable, and the cysts are not necessarily round (they may be bilobed or branching). The intervening lung parenchyma appears normal. In the late stage, there may be diffuse cysts, with no nodules evident (about 20% of cases [6]), while in the early stages, only nodules may be seen [3]. In the late stages, the disorder may be indistinguishable from lymphangiomyomatosis- but sparing of the costophrenic angles suggests the diagnosis of histiocytosis X [6]. Mediastinal adenopathy has been described in some series, but is usually uncommon [2]. Other authors report medistinal adenopathy in up to 30% of EG patients [10].
Scintigraphy: Gallium-67 scans are generally negative [12].
REFERENCES:
(1) Radiographics 1997; Mar 17 (2):377-390
(2) AJR 1997; Gilkeson RC, et al. Chest case of the day. Histiocytosis X. 169(1): 268, 273-4 (No abstract available)
(3) J Thorac Imaging 1996; 11: 1-26 (p.22)
(4) Radiology 1997; Siegelman SS. Taking the X out of Histiocytosis X. 204: 322-324. Review. (No abstract available)
(5) Radiology 1997; 204: 497-502
(6) AJR 1998; Bonelli FS, et al. Accuracy of high-resolution CT in diagnosing lung diseases. 170: 1507-1512
(7) Thorax 1998; Parums DV. Commentary: "Histiocytosis X". 53: 322-323 (No abstract available)
(8) Thorax 1998; Habib SB, et al. Recurrence of recipient Langerhans' cell histiocytosis following bilateral lung transplantation. 53: 323-325
(9) Thorax 1998; Gabbay E, et al. Recurrence of Langerhans' cell granulomatosis following lung transplantation. 53: 326-327
(10) Society of Thoracic Radiology Meeting 1999; Jensen EA, et al. Intrathoracic lymphadenopathy associated with pulmonary Langerhan's cell histiocytosis. Presented at scientific session III- paper 3.
(11) Societyof Thoracic Radiology Course Syllabus 1999; Swensen SJ. HRCT of diffuse lung disease. p. 203-236
(12) Thorax 2000; Tazi A, et al. Adult pulmonary Langerhans' cell histiocytosis. 55: 405-416 (No abstract available)
(13) N Engl J Med 2000; Vassallo R, et al. Pulmonary langerhans'-cell histocytosis. 343: 1969-1978 (No abstract available)
(14) Chest 1999; Mogulkoc N, et al. Pulmonary Langerhans' cell histiocytosis. Radiologic resolution following smoking cessation. 115: 1452-1455
(15) Radiology2001; Collins J, et al. Frequency and CT findings of recurrent disease after lung transplantation. 219: 503-509
(16) Chest 2004; Mendez JL, et al. Pneumothorax in pulmonary langerhans cell histiocytosis. 125: 1028-1032
(17) Radiographics 2004; Abbott GF, et al. Pulmonary langerhans cell histiocytosis. 24: 821-841
(18) Radiographics 2007; Leatherwood DL, et al. Pulmonary langerhans cell histiocytsis. 27: 265-268
(19) Radiographics 2008; Attili AK, et al. Smoking-related interstitial lung disease: radiologic-clinical-pathologic correlation. 28: 1383-1398