Top Story

Latest News


Sponsored
We care about what you think.
October 30, 2023

Deep-learning model determines breast tumor staging
April 16, 2024

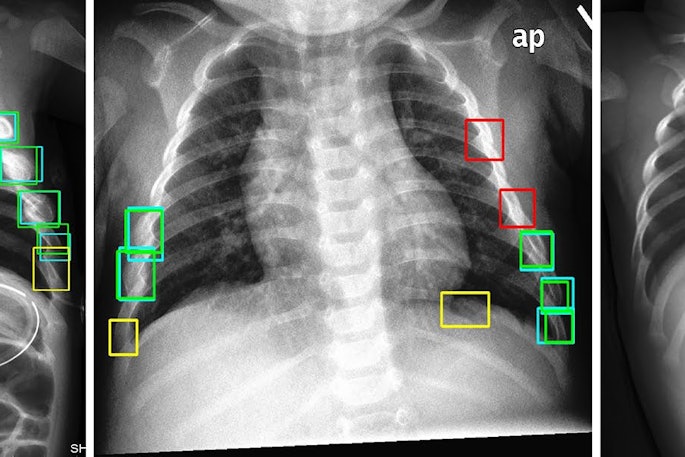
FAPI-PET shows promise in patients with lung cancer
April 15, 2024
Cases of the Week
Check out our Cases of the Week!




More from AuntMinnie

Matsumoto named chair of ACR Board of Chancellors
April 16, 2024

SNMMI declares win in Washington, DC
April 16, 2024

ACR lays off 11% of workforce
April 16, 2024
SIR publishes statement regarding pediatric trauma care
April 15, 2024


Flywheel CEO to step down
April 15, 2024
Sonio taps new scientific advisory board member
April 15, 2024



